
Abstract
Character states during sporulation have been used to segregate and describe many small-spored species of Alternaria, but some are not supported by published phylogenetic analyses. The conidiation response of Alternaria gaisen was characterized by selective subtractive hybridization of cDNA produced from cultures of A. gaisen grown either in total darkness or in total darkness followed by scarification and 24 h exposure to light. Transcripts or their translation products were identified using BLAST. Multiple transcripts with similarity to ORF-1 of the AM-toxin gene were obtained from the light library. L152 is a full reading frame EST in the light library whose ORF translation has similarity to the conserved domain aegerolysin (pfam06355). A set of 11 ex-type or representative isolates including A. alternata, A. gaisen, A. yaliinficiens, A. arborescens, A. tenuissima and A. brassicicola were resolved by UPGMA analysis of a partial genomic sequence (415–425 base pairs) of L152, but were not resolved by a similar analysis of ITS sequences. Furthermore, the resolved lineages of the L152 dataset were reflective of the diversity previously hypothesized by morphological evaluations of sporulation patterns. Although the ITS rDNA sequence region is generally accepted as the most likely candidate for fungal barcoding, the analysis of L152 sequences presented here resolved closely related species or species groups where other loci, including ITS, have not. Based on these results, the sequences of putative aegerolysin homologs were variable, parsimony-informative and warrant additional analyses with a broader isolate set including related genera and species.
Similar content being viewed by others

Introduction
Alternaria Nees is a cosmopolitan, anamorphic hyphomyceteous genus containing many species of economic importance, including saprophytes, phytopathogens, zoopathogens and as producers of mycotoxins and allergens. A recent class-level multigene phylogenetic analysis of the Dothideomycetes (Schoch et al. 2009) reaffirmed placement of Alternaria in the Pleosporales, a group heavily represented by phytopathogens. Disease reports and isolation records indicate discontinuous distribution of some phytopathogenic species of Alternaria (Farr and Rossman 2010; Simmons 2007) and in such cases these species may be considered exotic by an importing country and therefore subject to official regulation by that country (Anonymous 1995). The accuracy and effectiveness of phytosanitary regulatory activities are, from the outset, dependent upon correct identification of the pests of interest. In addition to being reflective of evolutionary history, a modern approach to taxonomy could include predictive biological information (e.g., biogeography, host ranges) that might not be reflected in some molecular analyses but be useful in managing phytosanitary issues (Roberts et al. 2000).
Alternaria taxonomy is discordant due to conflicting results from molecular and morphological analyses. Rossman (2007) described the rationale for selection of ITS for barcoding the fungi but acknowledged that ITS does not work for some clearly recognizable species, and Seifert (2009) noted that additional barcode genes may be necessary for such fungi. Molecular studies using ITS rDNA (Kusaba and Tsuge 1995), protein coding housekeeping genes (Peever et al. 2004), and anonymous open reading frames (ORF; Andrew et al. 2009) sequence data, rDNA RFLPs (Kusaba and Tsuge 1994), and IGS restriction mapping (Hong et al. 2005) from the small-spored Alternaria have demonstrated a low level of variability that do not support most segregations resulting from morphological, chemotaxonomic, and DNA fingerprinting studies. Kusaba and Tsuge (1995) went as far as to conclude that the small-spored species are all intraspecific variants of one species, A. alternata. However, species such as A. gaisen and A. yaliinficiens that, prior to 1992 and 1995, respectively, were geographically limited to Asia (Kohmoto et al. 1992; Baudry et al. 1993; Roberts 2005) are supported in DNA fingerprint analyses and metabolite profiles that have supported morphological segregation of several species-groups and are generally congruent with morphological character states (Roberts et al. 2000; Hong et al. 2005; Peever et al. 1999; Andersen and Thrane 1996; Andersen et al. 2001, 2002). We used subtractive hybridization to isolate and characterize sporulation-associated expressed sequence tags (ESTs) produced at the onset of conidial chain formation in response to light exposure, which we determined is required for conidiation in A. gaisen RGR91.0166 but not in A. alternata RGR91.0102 (data not shown). Stable and discriminative variation in sporulation patterns and spore characteristics have been observed among A. gaisen, A. longipes, and the small-spored Alternaria species from citrus and was shown to be important in elucidating natural history information, especially host-specific pathogenicity (Simmons 1999a). Hong et al. (2006) suggested that finding more informative molecular loci may be necessary to fully resolve the taxonomy of Alternaria and rectify it with the observed morphological distinctions.
Simmons’ methods (Simmons 2007) to grow and observe Alternaria for morphologic study prescribe growing them on low nutrient media under an 8/16-h cycle of fluorescent light and darkness. We reasoned that sampling mRNA populations during discriminative morphological development might provide access to previously unknown discriminative sequences for Alternaria. We initiated a search for candidate genes by noting the phenology of sporulation of Alternaria under controlled conditions, then used selective subtractive hybridization to identify and characterize mRNA transcripts harvested during early conidial chain formation. Here, we report the isolation and characterization of a set of differentially expressed transcripts from A. gaisen RGR91.0166 collected before and after 24 h exposure to light, and present a comparative preliminary UPGMA analysis based upon ITS and one class of transcripts with similarity to aegerolysin that are strongly upregulated by light exposure.
Materials and methods
Growth, observation and harvest of Alternaria isolates
Cultures used in this study are given in Table 1, and were maintained as agar blocks held in refrigerated water tubes. All cultures were grown and maintained on potato carrot agar (PCA) made by the method of Simmons (2007). Observations of conidiophore and conidium development were made using a stereomicroscope at ×50 magnification. For the dark-exposed RNA, A. gaisen was grown on Amersham Hybond-N nylon membranes on PCA plates inoculated with a mycelial agar plug and incubated in total darkness at 23°C and 21% RH. After 4 days, the mycelium was harvested by scraping and then suspended in liquid nitrogen. The plates were returned to the incubator and held for an additional 24 h but under continuous fluorescent light, then the conidiophores and conidia produced were scraped and processed as described.
RNA extraction
mRNA was extracted from crushed, frozen mycelium with a Qiagen RNeasy Plant mini-kit (Qiagen Sciences, MD, USA) per the manufacturer’s instructions. All RNA extracts were treated with RQ1 RNase-Free DNase (Promega, Madison, WI, USA) following the manufacturer’s instructions.
Observation of morphologic response
Cultures of A. gaisen were observed using stereomicroscopy at ×50 after both 4 days of incubation in the dark and again after scarification and an additional 24 h of growth under constant light. Cultural characters and developmental status of conidiophores and conidia were noted.
Selective subtractive hybridization (SSH)
All kits were used according to the manufacturer’s instructions except where noted. First strand cDNA was synthesized from total RNA from both the dark- and light-exposed tissues using the Super SMART PCR cDNA Synthesis Kit (Clontech, Mountain View, CA, USA) and purified using the Clontech NucleoSpin Extract II kit. First strand cDNA was amplified by long distance PCR and cleaned using a QIAquick PCR purification kit (Qiagen) in lieu of the phenol/chloroform cleanup protocol in the manufacturer’s instructions, then digested with RsaI (New England Biolabs, Ipswich, MA, USA). Differentially-expressed ESTs from A. gaisen were obtained by subtractive hybridization of cDNA using a Clontech PCR-Select cDNA Subtraction kit. All clones were screened for cDNA inserts by PCR using NewSP6 and T7 sequencing primers (Eurofins MWG Operon, Huntsville, AL, USA). A Clontech PCR-Select Differential Screening Kit and virtual Northern analysis were used to confirm the differential expression of ESTs (Table 3). A reverse subtraction, conducted as above but with tester and driver strains reversed, was also completed. ESTs with confirmed specificity were maintained in a light or dark cDNA library for A. gaisen.
cDNA insert-bearing plasmids were recovered using an Invitrogen S.N.A.P. Miniprep Kit (Invitrogen, Carlsbad, CA, USA) and sequenced using a Beckman GenomeLab Dye Terminator Cycle Sequencing Quick Start Kit (Beckman Coulter, Fullerton, CA, USA) with primers NewSP6 (Operon) and pUC/M13 forward (Promega) and analyzed using a Beckman CEQ8000 DNA sequencer. The forward and reverse sequences were screened for vector sequences then aligned and edited to give consensus sequences. GenBank was queried for homologous sequences in BLAST (Altschul et al. 1990) using blastn, blastx or ORF finder/blastp queries sequentially. Blastn searches of the nt/nr, est_others, and wgs databases were made in preferential order. Blastx queries of non-redundant protein (nr) sequences used the default nucleotide translation.
Analysis of L152 and ITS5
Primers L152-F1 and L152-R1 were designed from the distal bases of L152 (Table 2). The L152 primers were used in PCR reactions to produce amplicons from genomic DNA of a selected set of ex-type cultures or representative cultures representing sporulation pattern groups 2–5 Simmons (Simmons 1993; Table 1) and A. yaliinficiens (Roberts 2005). The L152 amplicons were sequenced as described, aligned and edited to 415–425 bp using default parameters in MUSCLE (Edgar 2004). ITS sequences obtained using ITS4 and ITS5 primers (White et al. 1990; Table 2) were aligned and trimmed to about 500 bp. Similarity matrices were calculated for L152 and ITS in BioNumerics 6.1 from which a UPGMA dendrogram was constructed for each sequence type. Analyses of L152 and ITS sequences used the default cost table, no correction and 0% gap penalty for alignments. Cluster analysis was by UPGMA, with 1,000 bootstrap analyses and 100% open gap cost, 0% unit gap cost, minimum match sequence=2, maximum number of gaps=9.
Hemolysis assay
Cultures of A. gaisen were grown in YG medium (Haffie et al. 1985) and incubated at 200g and 25C for 24–48 h. Mycelium was decanted through Miracloth, rinsed with distilled water, and then dehydrated in two changes of 200 ml of acetone. The acetone was decanted and the defatted mycelium was frozen, lyophilized, finely ground, then 1.9 g powdered mycelium was extracted with agitation for 24 h at 25°C in 100 ml modified (phenol omitted) Coca’s solution (NaHCO3, 3 g; NaCl, 9 g; phenol, 5 g; H20, 1 L) (Paris et al. 1990). The extract was centrifuged at 5,000g for 1 h at 4°C and the supernatant was collected, frozen and lyophilized in a tared centrifuge tube. The dry extract was reconstituted to a 10% extract by weight by addition of an appropriate volume of 0.05 M TRIS-HCl, pH 8.0. About 100 ml of reconstituted extract was placed into wells cut into sheep red blood cell agar (SRBC; Teknova, CA, USA) and the plates were held at 37°C for 24 h. A control well received only TRIS-HCL. A positive assay for hemolytic activity was determined as a zone of clearing around the point of application of the extract caused by lysis of the embedded red blood cells.
Results
Morphologic observations
Radial growth of dark-grown mycelium was ca 40 mm diam after 4 days. A sparse growth of funiculose hyphal elements developed vertically from the mycelium. Nearly all sporulation observed was directly upon the agar block and from aerial mycelium that grew from the plug. Conidia were in short, unbranched chains, generally 5 or less per chain. Conidiophores developed synchronously from the membrane surface after light exposure, nearly all of which exhibited a single, hyaline conidium, but occasionally chains of up to three conidia were present.
Subtractive hybridizations
Differentially-expressed transcripts in both libraries (Table 3) were most similar to sequences of related Dothideomycetous taxa; Pyrenophora tritici-repentis, Phaeosphaeria nodorum, and Cochliobolus heterostrophus and anamorphic Dothideomycetes. A total of 184 transcripts were differentially expressed and are listed by putative function in Table 3. Differential expression was confirmed by virtual northern analysis, but a small number were either not differentially expressed or were not expressed in the expected library. Of the transcripts, 72% returned either No Hit (29/184), best hits to whole genome shotgun sequences (WGS) with unknown functionality (35/184), or best hits to hypothetical proteins (68/184) with no putative function. Some ESTs present in both libraries but upregulated in the dark were similar to genes with conidiation-associated regulatory function (D282, D150, D188, L40). Other conidiation-associated ESTs (L152, L182, L3) were strongly upregulated after light exposure.
The most abundant class of transcripts showed similarity to a hypothetical protein in the AM-toxin gene cluster and was strongly upregulated after light exposure (Table 3). L126 represents 30 transcripts, each with about 100 bp that are highly similar to bases 4,959–4,860 of the “AMT genes region” (AB525200.1). Twenty-one additional transcripts represented by L217 are complimentary to the region spanned by L126. L172 is similar to a putative intron at positions 97,086–97,350 on the (−) strand of AB525200.1. No transcript in either library was similar to an AK-toxin gene or homolog, except some similarity was seen with the AF-toxin biosynthesis gene cluster (AB179766).
Eleven transcripts are represented by L152, which encodes a 402 bp (−1) reading frame whose translation product is a 133aa hypothetical protein with similarity to aegerolysin, a conserved domain (pfam06355). L152-F1 and -R1 primers gave a 445-bp product from genomic DNA of A. gaisen RGR91.0166.
Hemolysis assay
Hemolytic activity of Coca’s extracts of powdered mycelium from A. gaisen RGR91.0166 was confirmed by a zone of SRBC lysis ca 5 mm wide around the wells after 24 h at 37°C. The TRIS negative controls did not produce a zone of SRBC lysis.
Sequence analysis of L152 and ITS-5
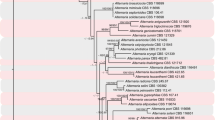
L152 primers produced a single amplicon from genomic DNA from 11 Alternaria isolates representing described sporulation phenotypes 2–5 (Table 1, Simmons 1993), A. yaliinficiens and A. brassicicola. A. arborescens and an unnamed arborescens-group isolate RGR91.0117 each produced two amplicons, the smaller of which was the expected size and was sequenced. Phylograms from the UPGMA analysis of ITS-5 and L152 are presented in Fig. 1, and the MUSCLE alignments are available for download at www.TreeBASE.org (SN10790; Sanderson et al. 1994). The ITS alignment revealed 0 parsimony informative characters and the analysis showed all ten representatives of groups 2–5 and A. yaliinficiens in a single unresolved clade. The alignment matrix of derivative L152 sequences contained 102 parsimony informative characters (25% of total characters) and resolved the small-spored species into two primary clades with 99% support. The uppermost clade resolved as three terminal clades corresponding to A. alternata, A. arborescens, and A. tenuissima, but with low bootstrap support for the A. tenuissima group. The lowermost clade resolved A. gaisen and A. yaliinficiens with high bootstrap support.

UPGMA analyses of ITS4-ITS5 (ITS) and a partial sequence (415–425 bp) of a hypothetical protein with similarity to aegerolysin (L152, GW574213). Numbers at nodes are bootstrap values from 1,000 replications. GenBank accession numbers for each sequence are given after the specific epithet or group designation
Discussion
Stable and discriminative phenotypic expression in small-spored Alternaria is most obvious in the morphologic variations of sporulation pattern and spore characters, and for A. gaisen, A. longipes and the small-spored Alternaria species from citrus, have been shown experimentally to be predictive of important biological characters, especially host-specific pathogenicity (Simmons 1999a, 1999b; Simmons and Roberts 1993). The synchronous conidiophores and conidia produced after light exposure were used to isolate and characterize sporulation-associated ESTs produced at the onset of conidial chain formation in response to light exposure, which we determined is required for conidiation in A. gaisen RGR91.0166 but not in A. alternata RGR91.0102 (data not shown).
Regarding the detection of transcripts with some similarity to AM-toxin synthetase, homologs of biosynthetic genes involved in toxin biosynthesis are thought to be shared among those pathotypes whose toxins share a common moiety (Tanaka et al. 1999; Masunaka et al. 2005) and AM-toxin is thought to be limited to a single lineage of ‘A. alternata apple pathotype’ (Johnson et al. 2000; Bushley and Turgeon 2010), not A. gaisen. AM-toxin and AK-toxin have different moieties, so the presence of a genomic sequence in A. gaisen with similarity to an AM-toxin gene was unexpected. We confirmed the absence of AK-toxin homologs or transcripts by PCR from genomic DNA of two isolates of ‘A. alternata apple pathotype’ and A. alternata, and confirmed the absence of AM-toxin transcripts in A. gaisen and A. alternata. AK-toxin production by A. gaisen RGR91.0166 was reported by Simmons and Roberts (1993) based upon excised leaf assays and confirmed by Roberts (2005) using PCR, who also reported the presence of AM-toxin PCR transcripts in two common reference strains of ‘A. alternata apple pathotype’, RGR87.0010 and RGR87.0031. None of the other isolates listed in Table 1 produced amplicons with primers for either AK- or AM-toxins except A. brassicicola, which was not tested (Roberts 2005). The detection of L126 PCR products from RGR87.0010 but not RGR87.0031, both of which are AM-toxin producers, is inconsistent with L126 being an AM-toxin homolog. AM-toxin synthetase is part of a large and diverse family of exclusively fungal non-ribosomal peptide synthetase (NRPS) genes with multiple domains (Bushley and Turgeon 2010). L126 may instead be an uncharacterized NRPS with partial similarity to one of the AM-toxin gene domains and expressed in response to conidiation-conducive stimuli. We did not detect L126-related sequences in A. alternata and one strain of ‘A. alternata apple pathotype’, so we do not consider this transcript a candidate for phylogenetic study.
Similarity of transcripts represented by L152 to the aegerolysin domain is notable because it is not represented amongst the unigenes occurring in the Pleosporales, even though the domain is conserved in bacteria and fungi. The distribution, function and role of fungal hemolysins were recently reviewed by Berne et al. (2009), who posited that aegerolysin was present in the ancestral lineage that gave rise to both Ascomycetes and Basidiomycetes. The predicted protein size from the L152 EST is similar to that of other fungal aegerolysins (Berne et al. 2009). Hemolytic activity of Coca’s extracts of A. gaisen RGR91.0166 was confirmed and demonstrates at least one of the functional associations for aegerolysin (0019835: cytolysis). Although L152 was first detected from the phytopathogen A. gaisen, the presence of putative aegerolysin homologs in both phytopathogenic and non-phytopathogenic species (Fig. 1) argues against its involvement in pathogenesis.
The sequence analysis of a putative aegerolysin homolog resolved several species of Alternaria that some have considered conspecific with A. alternata (Kusaba and Tsuge 1995; Peever et al. 2004). The relatively invariable ITS sequence for these species, which has been used to support the synonymy of A. gaisen and A. alternata, did not resolve these same isolates. With such a limited taxon sample we hesitate to draw broad conclusions or predictions about the utility and resolution of the putative aegerolysin from Alternaria for large-scale phylogenetic analyses. A complete genomic sequence of L152 and flanking regions may inform the design of competent sequencing primers and allow use of the putative aegerolysin L152 for expanded phylogenetic analyses, and efforts to obtain the complete L152 genomic sequence have begun. Even with such a limited isolate set, however, it is clear that even the partial-sequence analysis in this paper was parsimony-informative and provided well-supported segregation of A. alternata from several species or groups with which it is often considered conspecific in the current mycological literature. Furthermore, because we have detected the putative aegerolysin homolog from species such as A. yaliinficiens and A. mali (Roberts 1924) that do not produce the extracellular toxins associated with the various “pathotypes” of A. alternata, we deduce that the putative aegerolysin gene does not reside on one of the conditionally-disposable chromosomes known to carry the genes responsible and is therefore likely to be present in all small-spored Alternaria spp., not just the toxin producers.
Identifying genes expressed during discriminative developmental processes such as sporulation in Alternaria spp. did result in discovery of discriminative gene sequences such as L152, and is proof of concept that such discriminative sequences do exist in small-spored Alternaria spp. Such work should continue and be expanded, as there are surely other discriminative gene sequences to be discovered and utilized. Among the ESTs isolated in this study were several with similarity to genes with known regulatory function and have been functionally associated with sporulation; phosphoglucomutase (L3), Pka (protein kinase, D150), and a translation initiation inhibitor-like protein (L40). All should be considered for additional study. Additionally, inclusion of other developmental stages in time-course studies may reveal genes with different patterns of temporal expression such as hemolysin, of which there are apparently several forms that are expressed at different developmental stages (Pires et al. 2009).
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Andersen B, Thrane U (1996) Differentiation of Alternaria infectoria and Alternaria alternata based on morphology, metabolite profiles, and cultural characteristics. Can J Microbiol 42:685–689
Andersen B, Kroger E, Roberts RG (2001) Chemical and morphological segregation of Alternaria alternata, A. gaisen, and A. longipes. Mycol Res 105:291–299
Andersen B, Kroger E, Roberts RG (2002) Chemical and morphological segregation of Alternaria arborescens, A. infectoria and A. tenuissima species-groups. Mycol Res 106:170–182
Andrew M, Peever TL, Pryor BM (2009) An expanded multilocus phylogeny does not resolve morphological species within the small-spored Alternaria species complex. Mycologia 101:95–109
Anonymous (1995) Sanitary and phytosanitary measures In: Annex 1A of the Uruguay round agreement, World Trade Organization, Geneva. Available: http://www.wto.org/english/docs_e/legal_e/legal_e.htm#sanitary. Accessed 2010 Sep 21
Baudry A, Morzieres JP, Larue P (1993) First report of Japanese pear black spot caused by Alternaria kikuchiana in France. Plant Dis 77:428
Berne S, Ljerka L, Sepčic K (2009) Aegerolysins: structure, function, and putative biological role. Prot Sci 18:694–706
Bushley KE, Turgeon GB (2010) Phylogenomics reveals subfamilies of fungal nonribosomal peptide synthetases and their evolutionary relationships. BMC Evol Biol 10:26
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Farr DF, Rossman AY (2010) Fungal databases, systematic mycology and microbiology laboratory, ARS, USDA. Available: http://nt.ars-grin.gov/fungaldatabases/. Accessed 2010 Apr 15
Haffie TL, Louie PW, Khachatourians GG (1985) Isolation of noninhibitory strains of Zymomonas mobilis. Appl Environ Microbiol 49:1007–1009
Hong SG, Liu D, Pryor BM (2005) Restriction mapping of the IGS region in Alternaria spp. reveals variable and conserved domains. Mycol Res 109:87–95
Hong SG, Maccaroni M, Figuli PJ, Pryor BM, Bellisario A (2006) Polyphasic classification of Alternaria isolated from hazelnut and walnut fruit in Europe. Mycol Res 110:1290–1300
Johnson RD, Johnson L, Kohmoto K, Otani H, Lane CR et al (2000) A polymerase chain reaction-based method to specifically detect Alternaria alternata apple pathotype (A. mali), the causal agent of Alternaria blotch of apple. Phytopathology 90:973–976
Kohmoto K, Otani H, Cavanni P, Bugiani R (1992) Occurrence of the Japanese pear pathotype of Alternaria alternata in Japanese pear orchards in Italy. Phytopathol Mediterr 31:141–147
Kusaba M, Tsuge T (1994) Nuclear ribosomal DNA variation and pathogenic specialization in Alternaria fungi known to produce host-specific toxins. Appl Environ Microbiol 60:3055–3062
Kusaba M, Tsuge T (1995) Phylogeny of Alternaria fungi known to produce host-specific toxins on the basis of variation in internal transcribed spacers of ribosomal DNA. Curr Genet 28:491–498
Masunaka A, Otani K, Peever TL, Timmer T, Tsuge T et al (2005) An isolate of Alternaria alternata that is pathogenic to both Tangerines and Rough Lemon and produces two host-selective toxins, ACT- and ACR-toxins. Phytopathology 95:241–247
Paris S, Fitting C, Ramirez E, Latgé JP (1990) Comparison of different extraction methods of Alternaria allergens. J Allergy Clin Immunol 85:941–948
Peever TL, Canihos Y, Olsen L, Ibañez A, Liu Y-C et al (1999) Population genetic structure and host specificity of Alternaria spp. causing brown spot of Minneola tangelo and rough lemon in Florida. Phytopathology 89:851–860
Peever TL, Su G, Carpenter-Boggs L, Timmer LW (2004) Molecular systematics of citrus-associated Alternaria species. Mycologia 96:119–134
Pires ABL, Gramacho KP, Silva DC, Góes-Neto A, Silva MM, Muniz-Sobrinho JS, Porto RF, Vilella-Dias C, Brendel M, Cascardo JCM, Periera GAG (2009) Early development of Moniliophthora perniciosa basidiomata and developmentally regulated genes. BMC Microbiol 9:158
Roberts JW (1924) Morphological characters of Alternaria mali Roberts. J Agric Res 27:699–708
Roberts RG (2005) Alternaria yaliinficiens sp. nov. on Ya Li pear fruit: from interception to identification. Plant Dis 89:134–145
Roberts RG, Reymond ST, Andersen B (2000) RAPD fragment pattern analysis and morphological segregation of small-spored Alternaria species and species-groups. Mycol Res 104:151–160
Rossman AY (2007) Report of the planning workshop for all fungi DNA barcoding. Inoculum 58(6):1–5
Sanderson MJ, Donoghue MJ, Piel W, Eriksson T (1994) TreeBASE: a prototype database of phylogenetic analyses and an interactive tool for browsing the phylogeny of life. Am J Bot 81:183
Schoch CL, Crous PW, Groenewald JZ, Boehm EWA, Burgess TI et al (2009) A class-wide phylogenetic assessment of Dothideomycetes. Stud Mycol 64:1–15
Seifert KA (2009) Progress towards DNA barcoding of fungi. Mol Ecol Resour 9(Suppl 1):83–89
Simmons EG (1993) Alternaria themes and variations (63–72). XI. Alternaria gaisen and the black spot disease of Japanese pear. Mycotaxon 48:91–107
Simmons EG (1999a) Alternaria themes and variations (226–235). Classification of Citrus pathogens. Mycotaxon 70:263–323
Simmons EG (1999b) Alternaria themes and variations (236–243). Host-specific toxin producers. Mycotaxon 70:325–369
Simmons EG (2007) Alternaria: an identification manual. CBS biodiversity series no. 6. CBS Fungal Biodiversity Centre, Utrecht
Simmons EG, Roberts RG (1993) Alternaria themes and variations (73). XII. Morphology and toxigenicity of Alternaria associated with black spot disease of Japanese pear. Mycotaxon 48:109–140
Tanaka A, Shiotani H, Yamamoto M, Tsuge T (1999) Insertional mutagenesis and cloning of the genes required for biosynthesis of the host-specific AK-toxin in the Japanese pear Pathotype of Alternaria alternata. Mol Plant Microb Interact 12:691–702
White TJ, Bruns TD, Lee S, Taylor JW (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand D, Sninsky JS, White TJ (eds) PCR protocols: a guide to methods and applications, Academic, San Diego, pp 315–322
Acknowledgements
The authors wish to thank Pedro Crous and Birgitte Andersen for their constructive reviews of the manuscript, and to Emory Simmons for helpful comments and suggestions throughout the study.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Roberts, R.G., Bischoff, J.F. & Reymond, S.T. Differential gene expression in Alternaria gaisen exposed to dark and light. Mycol Progress 11, 373–382 (2012). https://doi.org/10.1007/s11557-011-0752-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11557-011-0752-3