
Cytosporin Derivatives from Arctic-Derived Fungus Eutypella sp. D-1 via the OSMAC Approach
by
 , ,
, ,
Hao-Bing Yu
1,†,
Zhe Ning
1,†,
Bo Hu
1,
Yu-Ping Zhu
2,
Xiao-Ling Lu
3,
Ying He
1,
Bing-Hua Jiao
3 and
Xiao-Yu Liu
1,* 1
Naval Medical Center of PLA, Department of Marine Biomedicine and Polar Medicine, Naval Medical University, Shanghai 200433, China
2
Basic Medical Experimental Teaching Center, College of Basic Medical Sciences, Naval Medical University, Shanghai 200433, China
3
Department of Biochemistry and Molecular Biology, College of Basic Medical Sciences, Naval Medical University, Shanghai 200433, China
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Mar. Drugs 2023, 21(7), 382; https://doi.org/10.3390/md21070382
Submission received: 14 June 2023
/
Revised: 26 June 2023
/
Accepted: 27 June 2023
/
Published: 28 June 2023
(This article belongs to the Special Issue Diversity of Marine Fungi as a Source of Bioactive Natural Products)
Abstract
:A chemical investigation of the Arctic-derived fungus Eutypella sp. D-1 based on the OSMAC (one strain many compounds) approach resulted in the isolation of five cytosporin polyketides (compounds 1–3 and 11–12) from rice medium and eight cytosporins (compounds 2 and 4–11) from solid defined medium. The structures of the seven new compounds, eutypelleudesmane A (1), cytosporin Y (2), cytosporin Z (3), cytosporin Y1 (4), cytosporin Y2 (5), cytosporin Y3 (6), and cytosporin E1 (7), were elucidated by analyzing their detailed spectroscopic data. Structurally, cytosporin Y1 (4) may be a key intermediate in the biosynthesis of the isolated cytosporins, rather than an end product. Compound 1 contained a unique skeleton formed by the ester linkage of two moieties, cytosporin F (12) and the eudesmane-type sesquiterpene dihydroalanto glycol. Additionally, the occurrence of cyclic carbonate moieties in compounds 6 and 7 was found to be rare in nature. The antibacterial, immunosuppressive, and cytotoxic activities of all compounds derived from Eutypella sp. D-1 were evaluated. Unfortunately, only compounds 3, 6, 8, and 10–11 displayed immunosuppressive activity, with inhibitory rates of 62.9%, 59.5%, 67.8%, 55.8%, and 68.7%, respectively, at a concentration of 5 μg/mL.
1. Introduction
Fungi have proven to be a valuable source of new secondary metabolites with a wide spectrum of biological activities [1]. Natural products from Polar fungi remain the non-negligible sources of pharmacologically active compounds [1]. Cytosporins are a family of hexahydrobenzopyran metabolites derived from fungi with a distinct heptene side chain residue [2]. Initially isolated from endophytic Cytospora sp. in 1996, cytosporins were recognized as inhibitors of angiotensin II binding inhibitors [2]. To date, nearly 30 natural cytosporins of this structural class have been predominantly isolated from four genera of fungi: Cytospora sp. [2], Pestalotiopsis sp. [3], Eutypella sp. [4,5], and Pseudopestalotiopsis sp. [6]. The cytosporin family exhibits diverse bioactive effects, including cytotoxic, antibacterial, and antagonistic activity [2,4]. Besides cytosporins, Eutypella species have been extensively investigated as a rich source of various bioactive compounds, pimarane diterpenes, γ-lactones, benzopyrans, ent-eudesmanes, cytochalasins, and dipeptides, which display a spectrum of bioactivities [7,8,9].
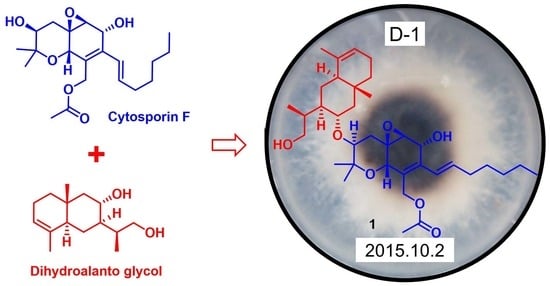
The OSMAC approach has emerged as a powerful tool in the field of natural product biodiscovery, stimulating the production of a wider range of new metabolites [10]. During our exploration of structurally diverse bioactive natural products from polar fungi, we discovered a series of terpenoids with unique skeleton characteristics from the talented Arctic fungus strain Eutypella sp. D-1 [5,7,9]. This strain has proven to be a prolific source of metabolites with diverse biological activities [7,9]. To enhance the chemical diversity of Eutypella sp. D-1, we employed the one strain many compounds (OSMAC) strategy, utilizing different culture conditions. Through high-performance liquid chromatography (HPLC) analysis, some structural analogs during fermentation on two distinct media, solid rice medium and defined solid medium, were dramatically discovered. Subsequent chemical investigation led to the isolation of 12 cytosporin derivatives, including seven new cytosporins—eutypelleudesmane A (1), cytosporin Y (2), cytosporin Z (3), cytosporin Y1 (4), cytosporin Y2 (5), cytosporin Y3 (6), and cytosporin E1 (7)—together with five known biogenetic-related analogs—cytosporin X (8), cytosporin E (9), cytosporin L (10), and cytosporins D and F (11–12) (Figure 1). Herein, we present the detailed purification, structure elucidation, and bioactive evaluation of these compounds.
2. Results
Eutypelleudesmane A (1) was isolated as a light-brown oil. The molecular formula was determined as C36H56O7 from HRESIMS and NMR data (Table 1), indicating the presence of nine degrees of unsaturation. The IR spectra confirmed the presence of hydroxy (3357 cm−1) and carbonyl (1741 cm−1) groups [3,4,5]. Additionally, the 13C NMR analysis revealed one ester carbonyl signal (δC 171.0) and six double-bond carbon signals (δC 121.2, 124.7, 125.1, 134.1, 135.6, and 136.4), accounting for four degrees of unsaturation. The remaining five degrees of unsaturation were attributed to the pentacyclic ring structure present in the molecule.
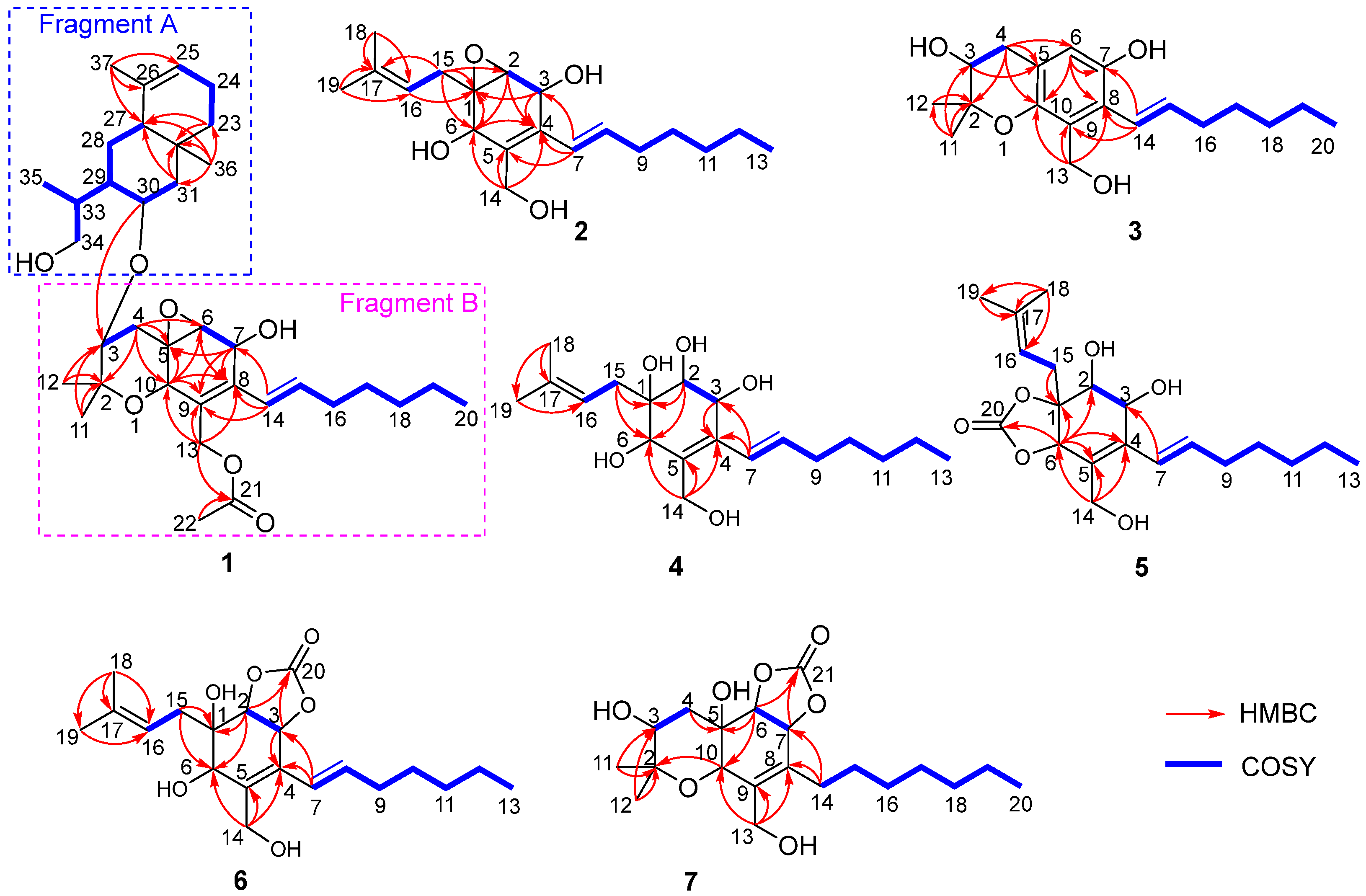
Upon comparing the 1D NMR data of compound 1 and cytosporin F (12), it was observed that one set of signals was similar to compound 12, while the remaining signals resembled a derivative of eudesmane-type sesquiterpene, dihydroalanto glycol. By utilizing 2D NMR correlations (Figure 2), these two structural fragments, labeled as A and B, were deduced. The COSY spectrum revealed the presence of seven continuous spin systems: (a) C-3−C-4, (b) C-6−C-7, (c) C-14–C-15–C-16–C-17–C-18–C-19–C-20, (d) C-23–C-24–C-25, (e) C-27–C-28–C-29–C-30–C-31, (f) C-29–C-33–C-34, and (g) C-33–C-35 (Figure 2). Fragment A, comprising C-2 to C-22, exhibited similarity to compound 12 based on a comparison of their 1D NMR spectra. HMBC correlations from H-4α to C-2, C-5, C-6, and C-10; from H-6 to C-8; from H-7 to C-5, C-8, and C-9; from H-10 to C-5, C-6, C-8, and C-9; from H3-11 and H3-12 to C-2 and C-3; and from H2-13 to C-8, C-9, and C-10 were detected. These correlations, along with the chemical shift of C-2 (δC 76.6) and C-10 (δC 67.5), indicated the formation of two six-membered rings by connecting C-5 (δC 55.7) with C-10 and C-2 with C-10 via an O-atom, as well as the location of the two methyl groups CH3-11 and CH3-12 both at C-2 and one methylene group CH2-13 at C-9. The presence of an oxirane resulting from the conjugation of C-5 and C-6 via O-atom was supported by the downfield shift of C-5 and C-6 (δC 59.7) [3,4]. Furthermore, the direct linkage between C-8 and C-14 was established by HMBC correlations from H-14 to C-7, C-8, and C-9. An additional acetyl group was identified to be connected to C-13 based on the HMBC correlations from H-13 and H-22 to C-21 and the chemical shift of C-13 (δC 61.5). Fragment B, spanning from C-23 to C-37, exhibited characteristics of a eudesmane-type sesquiterpene moiety, as deduced from the analysis of the remaining 1H and 13C NMR data. HMBC correlations from H-31α and H-31β to C-27 and C-32; from H2-23 to C-27 and C-32; and from H3-36 to C-23, C-27, C-31, and C-32 confirmed the presence of a linkage of C-23, C-27, and C-31 via the quaternary carbon C-32, and placed the methyl group CH3-36 at C-32 as well. The methyl group CH3-37 was demonstrated to be connected to C-25 and C-27 via C-26 by the HMBC correlations from H3-37 to C-25, C-26, and C-27. The linkage of fragments A with B through C-3 (δC 73.8) and C-30 (δC 66.7) via an O-atom was supported by the downfield resonance of C-3 and C-30, along with HMBC correlations from H-30 to C-3. Additionally, the connection of two hydroxyl groups with a downfield carbon shift at C-7 (δC 64.6) and C-34 (δC 67.5) were determined to satisfy the molecular formula. Consequently, the planar structure of 1 was established as depicted.
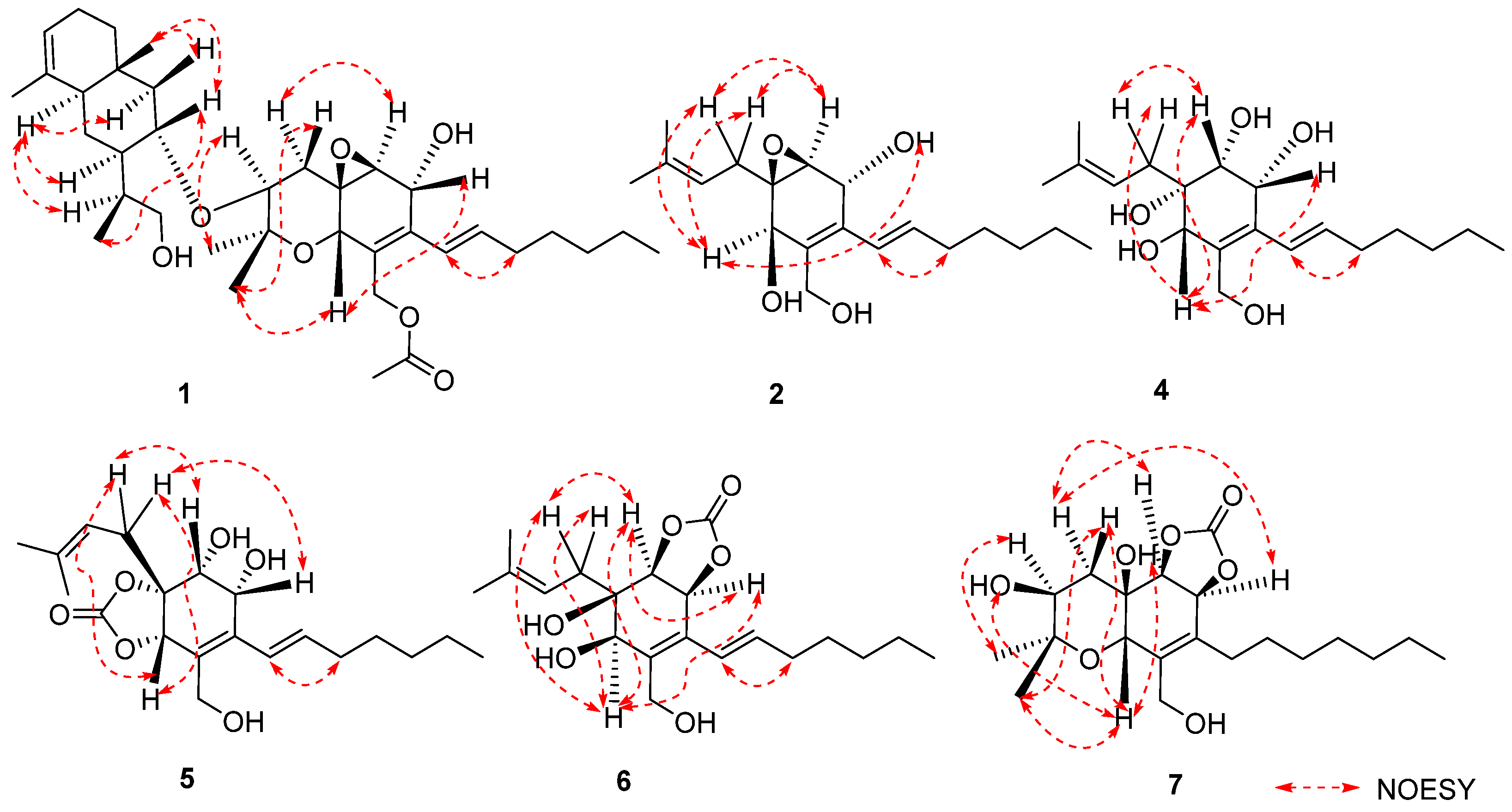
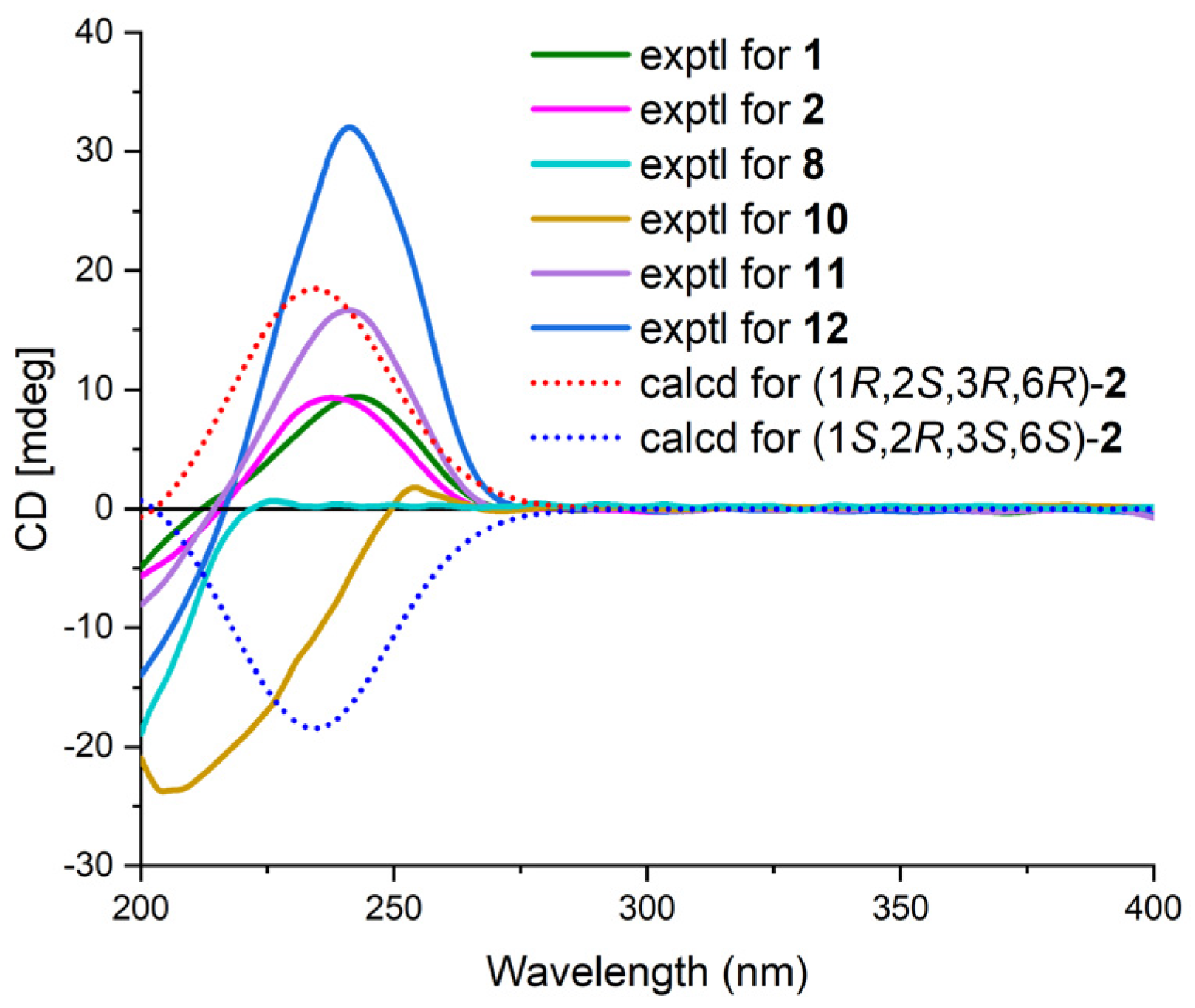
The relative configuration of 1 was established by analyzing coupling constants and NOESY experiments [9]. The trans configuration of the conjugated C-14/C-15 double bond was inferred based on the large coupling constant (16.0 Hz) and the NOESY correlations of H-14/H2-16. The observed similarity in the NMR chemical shift values and NOESY correlations of H-7/H-10, H-10/H3-12, H3-12/H-4β, H-4α/H-6, and H-3/H3-11 indicated that the relative configurations of fragment A in 1 were identical to those of 12 [3,4,5]. Additional NOESY correlations of H-30/H3-35, H-30/H-36β, and H-31β/H-36β and those of H-27/H-29, H-27/H-31α, and H-27/H-33 indicated the β-orientation and α-orientation of these protons in fragment B, respectively (Figure 3). Furthermore, the absence of a NOESY correlation between H-3 and H-30 supported the trans relationship between these two protons [5]. Thus, the relative structure of 1 was determined. Furthermore, the characteristic positive Cotton effect at 242 nm in the CD spectrum of 1 was virtually identical to that of cytosporins D and F (11–12) (Figure 4) [5], which suggested the absolute configuration of 1 was assigned as 3S,5R,6S,7R,10S,27S,29S,30R,32S,33R.
Cytosporin X (2) was obtained as a light-brown oil and determined to have a molecular formula of C19H30O4 based on HRESIMS and NMR data, corresponding to an unsaturation index of 5. The presence of hydroxy functionality was indicated by IR absorption bands at 3359 cm−1. The 13C NMR (Table 2) and DEPT spectra revealed the presence of 19 carbons, including six double-bond carbon signals (δC 117.3, 124.6, 131.4, 131.6, 135.4, and 135.9) and five oxygenated carbon signals (δC 57.4, 59.3, 62.2, 64.3, and 69.5). The COSY spectrum of 2 showed three distinct spin systems: C-2/C-3, C-7/C-8/C-9/C-10/C-11/C-12/C-13, and C-15/C-16 (Figure 2). HMBC correlations from H-2 to C-4 and C-6; from H-3 to C-1 and C-4; from H-6 to C-1, C-2, and C-4; and from H2-14 to C-4, C-5, and C-6, along with the comparison of the chemical shifts of C-1 (δC 59.3) and C-2 (δC 57.5) to those of cytosporins D and F [3,4], determined the oxirane-fused cyclohexene moiety with one methylene group (CH2-14) attached at C-5. The isoamylene group was connected to C-1 based on the HMBC correlations from H2-15 to C-1, C-2, and C-6, as well as from H3-18 and H3-19 to C-16 and C-17. Further HMBC correlations from H-7 to C-3, C-4, and C-5 established the connectivity of C-4 and C-7. With this assignment secured, each of the three oxygenated carbon at C-3 (δC 64.3), C-6 (δC 69.5), and C-14 (δC 62.2) had to be substituted with a hydroxy group to satisfy the molecular formula. The relative stereocenter of 2 was determined from NOESY correlations and coupling constants in comparison with those of 11 and 12 [3,4]. The conjugated C-7/C-8 double bond was assigned as trans upon its large coupling constant (16.0 Hz). The NOESY correlations of H-2/H2-15 and H-6/H2-15 in CDCl3 and 3-OH/H-6 in DMSO-d6 (Figure S16), combined with the similarity between the calculated and the experimental ECD spectra, confirmed the absolute configurations of 2 as 1R,2S,3R,6R (Figure 4).
Cytosporin Y (3) exhibited a negative HRESIMS with a pseudomolecular ion at m/z 319.1912 [M − H]−, consistent with the molecular formula of C19H28O4. The similarity of the 1H and 13C NMR data between 3 and 11 indicated that compound 3 was the derivative of 11. The presence of a pentasubstituted benzene moiety (δH 6.61 (1H, s); δC 115.1 (CH), 118.8 (C), 123.6 (CH), 126.8 (C), 144.8 (C), and 146.7 (C)) instead of the oxirane-fused cyclohexene moiety in 11 was suggested by the 1H and 13C NMR spectra. This was further confirmed by the further HMBC correlations from H-4β to C-5, C-6, and C-10; from H-6 to C-7, C-8, and C-10; from H2-13 to C-8, C-9, and C-10; and from H-14 to C-7, C-8, and C-9 (Figure 2). Additionally, one hydroxy group was attached to C-7, as evidenced by its chemical shift (δC 146.7) and the molecular formula. The conjugated C-14/C-15 double bond in 3 was assigned as trans based on the similar 1H NMR chemical shift values and coupling constants (16.5 Hz) observed in 3 and 2. To determine the absolute configuration at C-3 in compound 3, the specific rotation ( +12.3, MeOH, c 0.1 and +1.9, CDCl3, c 0.1) was measured. The configuration of C-3 could be assigned as S by comparison to the literature data for synthetic (R)-2,2-dimethylchromane-3,7-diol ( −1.2, MeOH, c 0.03) [11] and (S)-2,2-dimethylchromane-3,7-diol ( +11.5, CHCl3, c 1.0) [12] (differences in measured versus literature values likely stem from the different concentration and solvent).
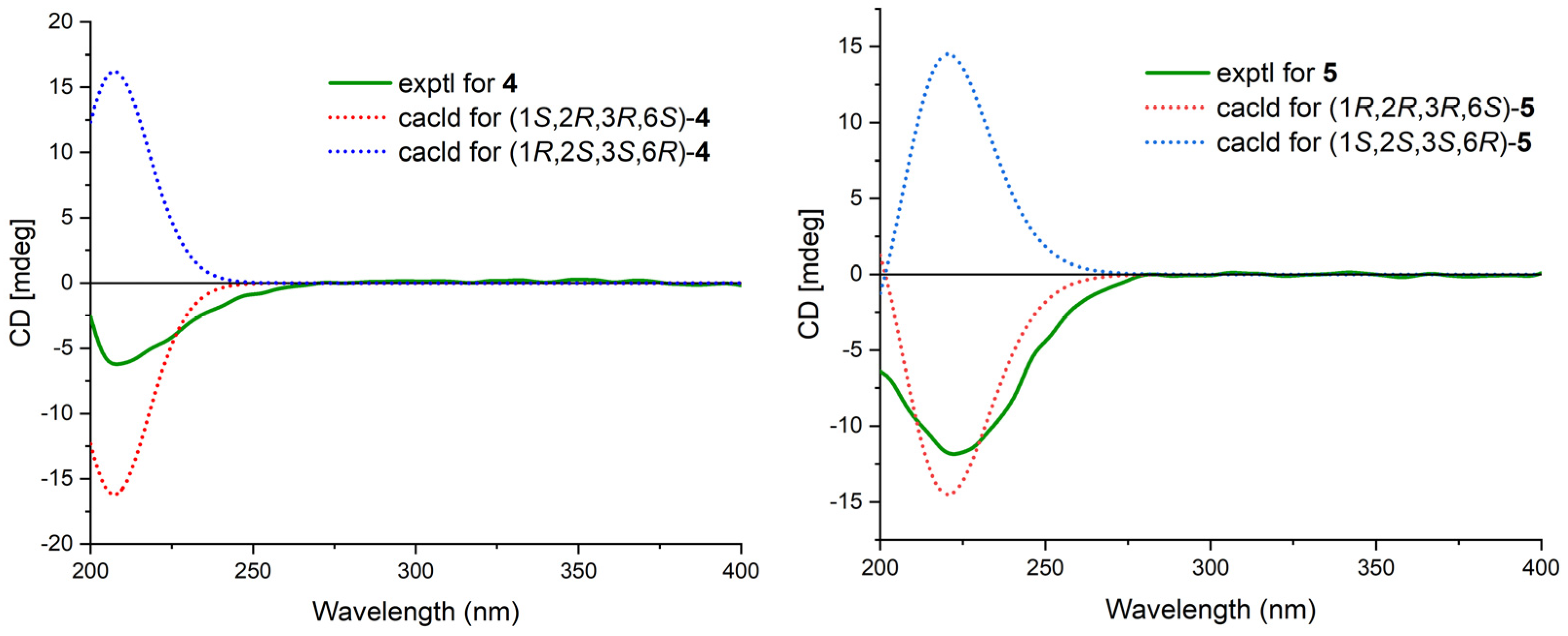
Cytosporin Y1 (4), in the form of a light-yellow oil, had a molecular formula of C19H32O5 based on HRESIMS (m/z 363.2139 [M + Na]+), which is larger than that of cytosporin Y (2) by 18 amu. The NMR data of 4 (Table 3) were nearly identical to those of 2, indicating the same carbon skeleton. Considering the degrees of unsaturation of 4, the observed downfield shift of one quaternary carbon (δC 59.3) and one methine (δH/δC 3.29/57.5) in 2 to δC 74.3 and δH/δC 3.76/75.0 in 4, respectively, suggested that 4 was the oxirane ring-opening product of 2. This hypothesis was further supported by further COSY and key HMBC correlations, as shown in Figure 2. The E-geometry of the Δ7,8 double bond was deduced from a NOESY correlation between H-7 and H2-9, as well as the coupling constants (16.5 Hz). Additional NOESY correlations of H-2/H2-15, H-2/H-6, H-3/H-6, and H-6/H2-15 indicated the same orientation of these protons. Furthermore, the comparison of the calculated and the experimental ECD spectra confirmed the absolute configurations of 4 as 1S,2R,3R,6S (Figure 5).
Cytosporin Y2 (5) was obtained as a light-yellow oil. Extensive NMR analyses and HRESIMS data (m/z 389.1928 [M + Na]+) led to the determination of its molecular formula as C20H30O6. The overall NMR data of 5 indicated a structure similar to 4, with the notable difference of an additional quaternary carbon. This carbon was identified as a carbonate moiety based on the strong IR absorption at 1647 cm−1 and the diagnostic 13C NMR signal at δC 154.8 [4]. Another significant difference was observed for C-1 and C-2, resonating at δC 74.3 and 75.0 in compound 4, whereas in compound 5, these signals resonated at δC 84.4 and 71.3, respectively (Table 3). This observation, along with the key HMBC correlations from H-2 to C-20, led to the linkage of the carbonyl to both oxygen atoms at C-1 and C-2 to form a cyclic carbonate moiety. The relative configurations of 5 were determined via a detailed analysis of the NOESY correlations of H-2/H-15a, H-3/H-15b, H-6/H-15a, H-6/H-15b, and H-7/H2-9, as well as the coupling constants (16.5 Hz) of H-7/H2-9. Furthermore, the calculated ECD spectrum of 5 exhibited a close resemblance to the experimental one, confirming the absolute configuration as 1R,2R,3R,6S (Figure 5).
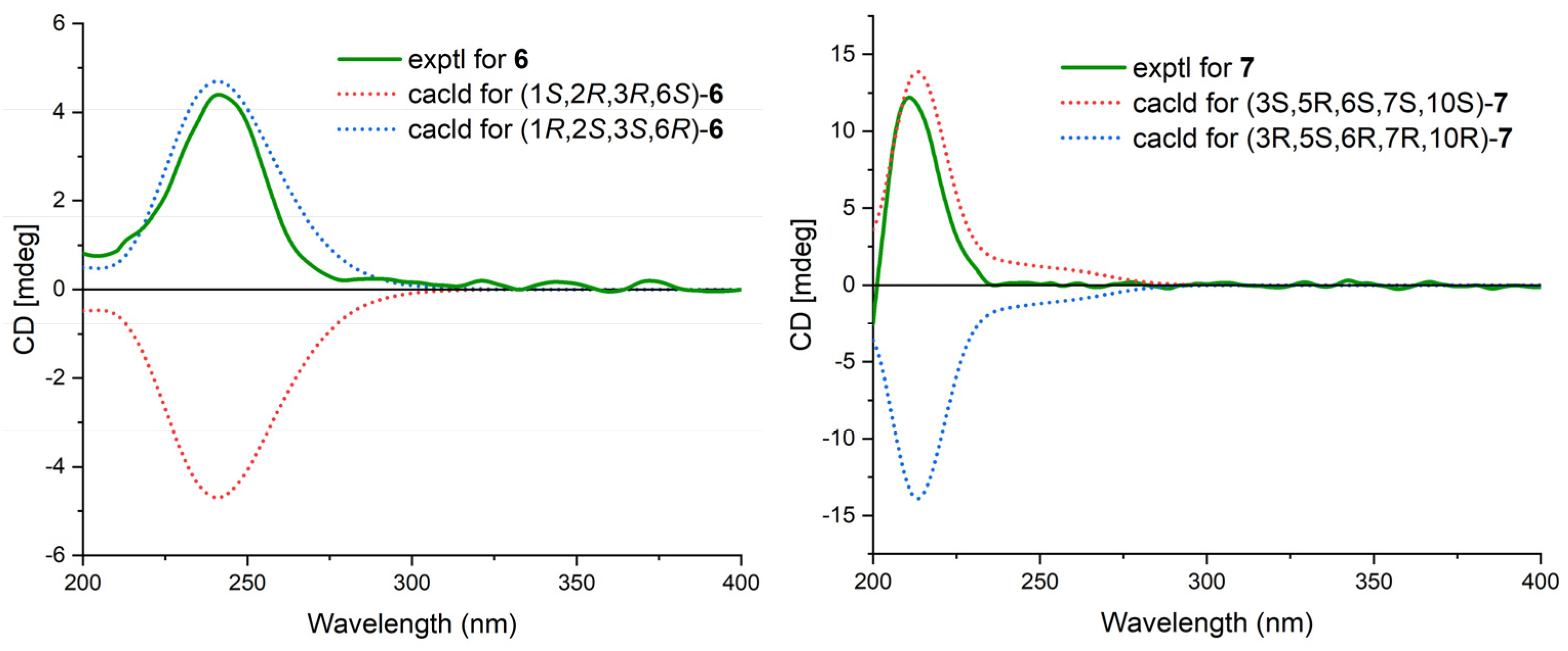
Cytosporin Y3 (6) was isolated as a light-yellow oil. Its molecular formula was determined to be C20H30O6, the same as that of 5, based on HRESIMS data. A comparison of the IR, UV, and NMR data (Table 4) of 6 with those of 5 suggested that 6 was an isomer of 5. Further analysis of the 13C NMR chemical shift of C-2 (δC 79.2) and C-3 (δC 75.2), along with the unambiguous HMBC correlations from H-2 and H-3 to C-20 of 2, revealed that the cyclic carbonate moiety was fused with C-2-C-3 in 6. The relative configuration and E-geometry of the Δ7,8 double bond in 6 were determined from the NOESY correlations of H-2/H-3, H-2/H-6, H-3/H-6, H-2/H-15b, H-6/H-15a, H-6/H-15b, and H-7/H2-9. The absolute configuration of 6 was subsequently determined to be 1R,2S,3S,6R based on the opposite CD spectra (Figure 6) and a comparison of the specific rotation ( +37.8, MeOH, c 0.1) with that of 4 ( −15.8, MeOH, c 0.1) and 5 ( −60.1, MeOH, c 0.1) (Figure 6).
Cytosporin E1 (7) was also purified as a light-yellow oil and exhibited a HRESIMS ion peak at m/z 407.2034 [M + Na]+, consistent with the molecular formula C20H32O7 with five degrees of unsaturation. The 1H and 13C NMR data of 7 (Table 4) closely resembled those of the known compound cytosporin E (9), except for two additional methylenes (δC/δH 31.0/2.26 and 30.0/1.49) in 7 and the absence of two olefinic methines (δC/δH 123.5/6.39 and 137.1/6.03) in 9. These observations indicated that C-8 in 7 was substituted by a heptane subunit instead of the 1-heptene part in 9, which was also confirmed by COSY correlations of H2-14 (δH 2.26)/H2-15 (δH 1.49), H2-15/H2-16 (δH 2.25), H2-16/H2-17 (δH 1.35), H2-17/H2-18 (δH 1.31), H2-18/H2-19 (δH 1.32), and H2-19/H3-20 (δH 0.90), as well as HMBC correlations from H2-14 to C-7 (δC 78.5) and C-8 (δC 133.3). The relative configuration of 7 was inferred to be different from that of compound 9 through a comparison of the 13C NMR data between 7 (C-6 δC 81.3 and C-7 δC 78.5) and 9 (C-6 δC 81.0 and C-7 δC 75.3), as well as the analysis of NOESY correlations of H-3/H3-11, H-4α/H-6, H-4α/H-7, H-4β/H-10, H-4β/H3-12, and H-10/H3-12 in MeOD-d4 and 3-OH/H-10 and 5-OH/H-10 in DMSO-d6 (Figure S75). The absolute configurations of 7 were subsequently assigned as 3S,5R,6S,7S,10S based on the similarity of its calculated and the experimental ECD spectra (Figure 6).
In addition to the seven new compounds 1–7, the five known cytosporins—cytosporin X (8) [13], cytosporin E (9) [4], cytosporin L (10) [14], cytosporin D (11) [4], and cytosporin F (12) [3]—were also isolated and identified through a comparison of its NMR spectroscopic data with reported values in the literature.
Structurally, considering the close relationship in biosynthesis among compounds 1–12, a biosynthetic pathway different from the previous literature for these compounds is proposed (Figure 7) [3]. The possible precursor originated from phenylmethanol [3]. The subsequent addition of an isoprenyl unit, followed by hydroxylation and the addition of an aliphatic chain, would give the intermediate i. The hydroxylation of the C-1/C-6 double bond in i gave rise to the key intermediate 4. Compound 2 was derived from 4 via a dehydration cyclization reaction. Compound 3 was generated from i via the epoxidation of the C-16/C-17 double bond and a cyclization reaction. Compounds 5 and 6 were derived from the dehydration reaction of compound 4 with carbonic acid by different attack directions and substitution positions, respectively. Compounds 7, 10, and 11 were obtained from 6, 4, and 2 via the same cyclization reaction as 3, respectively. Compound 9 was derived from 10 via the carbonic acid substitution, while compound 8 was formed through the hydrogenation of 11. The cyclization of compound 2, followed by an acetylation reaction, resulted in the formation of compound 12. Another possible precursor, the eudesmane-type sesquiterpene dihydroalanto glycol, was generated from farnesyl pyrophosphate with two steps of cyclization, dehydrogenation, and hydroxylation reaction [15]. Then, 1 was formed from the above two precursors, ii and 12, via a condensation reaction.
All the isolated compounds 1–12 were evaluated for their cytotoxicity against four human cancer cell lines, including DU145, SW1990, Huh7, and PANC-1, and antibacterial activity against Staphylococcus aureus, Escherichia coli, and Bacillus subtilis. Unfortunately, all compounds were not active during the above test, with IC50 values higher than 50 μM or MIC values higher than 128 μg/mL. Additional immunosuppressive activity against ConA-induced T cell proliferation for 1–12 was also tested. However, only compounds 3, 6, 8, and 10–11 displayed immunosuppressive activity, demonstrating inhibitory rates of 62.9%, 59.5%, 67.8%, 55.8%, and 68.7%, respectively, at a concentration of 5 μg/mL.
3. Materials and Methods
3.1. General Experimental Procedures
Specific rotations and IR (KBr) data were measured on a PerkinElmer model 341 polarimeter (Perkin-Elmer Inc., Waltham, MA, USA) and Jasco FTIR400 spectrometer (Jasco Inc., Tokyo, Japan), respectively. CD and UV spectra were obtained on a Jasco J-715 spectropolarimeter (Jasco Inc., Tokyo, Japan) and UV-8000 spectrophotometer (Shanghai Metash instruments Co., Shanghai, China) in MeOH, respectively. 1D and 2D NMR spectra were acquired using a Bruker AMX-500 instrument (500 MHz for 1H NMR, 125 MHz for 13C NMR) (Bruker Biospin Corp., Billerica, MA, USA) at room temperature. HRESIMS data were measured on an Agilent 6210 LC/MSD TOF mass spectrometer (Agilent Technologies Inc. Lake Forest, CA, USA). HPLC separation was performed using a YMC-Pack Pro C18 (5 μm) column (YMC Co. Ltd., Kyoto, Japan) using a Waters 1525 separation module equipped with a Waters 996 Photodiode Array (PDA) detector (Waters Corp., Milford, MA, USA). Column chromatographic purifications were performed on silica gel 60 (200–300 mesh, Qingdao Ocean Chemical Co., Qingdao, China), ODS (50 μm, YMC Co. Ltd., Kyoto, Japan), and Sephadex LH-20 (Pharmacia Co., Piscataway, NJ, USA).
3.2. Fungal Material
The fungus Eutypella sp. D-1 (GenBank accession number FJ430580) was separated from the sample collected near London Island of Kongsfjorden in the Ny-Ålesund District of the Arctic area and recognized based on 18S rDNA gene sequence analysis. The strain (No. D-1) was deposited in the Department of Marine Biomedicine and Polar Medicine, Naval Medical Center of PLA, Naval Medical University.
3.3. Fermentation, Extraction, and Isolation
The fungal strain Eutypella sp. D-1 was cultivated in seed medium (PDB 100 mL) in 250 mL Erlenmeyer flasks on a rotatory shaker (180 rpm) at 20 °C for 3 days. Subsequently, seed medium (10 mL) was transferred into 60 × 250 mL Erlenmeyer flasks (40 g of rice and 60 mL of water) and 60 plates of about 20 cm diameter (sucrose 51.4 g, NaNO3 3.3 g, K2HPO4·3H2O 0.07 g, MgSO4·7H2O 0.4 g, KCl 0.625 g, yeast extract 0.7 g, CoCl2·6H2O 0.003125 g, FeSO4 0.01875 g, CaCl2 0.0065 g, and l-ornithine hydrochloride 15 g, and agar 20.0 g, dissolved in 1 L of water), respectively, and then cultured under static conditions at 20 °C for 45 days.
The rice fermentation was combined and then extracted with CH2Cl2−MeOH (1:1, 1 L) three times. The organic solvent was concentrated under reduced pressure and partitioned with EtOAc and H2O to yield the EtOAc extract (24.5 g). The EtOAc extract was subjected to vacuum liquid chromatography (VLC) on silica gel via gradient elution using CH2Cl2/MeOH (80:1, 60:1, 40:1, 20:1, 15:1, 10:1, 0:1, v/v) as the solvent to give seven fractions (A–G). Fraction B (1.23 g) was chromatographed on a Sephadex LH-20 column using CH2Cl2−MeOH (1:1) as mobile phase to afford three subfractions (Fr. B1−B3), and subfraction B1 was further purified by reversed-phase HPLC eluting 43% MeCN/H2O at a flow rate of 2 mL/min to afford 1 (3.2 mg, tR = 16.6 min). Compounds 2 (7.3 mg, tR = 23.4 min) and 3 (1.4 mg, tR = 46.6 min) were isolated using reversed-phase HPLC (63% MeOH/H2O) from subfraction B2. Fraction F (3.75 g) was separated using MPLC on an ODS (50 μm) column to give seven fractions (Fr. F1–F7). Fr. F3 was subjected to reversed-phase HPLC (65% MeOH/H2O, 2 mL/min) to afford 11 (15.3 mg, tR = 15.3 min). Fr. F4 was separated with reversed-phase HPLC (40% CH3CN/H2O, 2 mL/min) to give 12 (40.1 mg, tR = 11.3 min).
The defined medium fermentation was combined and then extracted with CH2Cl2−MeOH (1:1, 1 L) three times. The organic solvent was concentrated under reduced pressure to yield the extract (6.86 g). The extract was subjected to silica gel VLC, eluting with a gradient of petroleum ether/EtOAc (100:1, 80:1, 50:1, 30:1, 20:1, 10:1, 5:1, 3:1, 2:1, 1:1, v/v) to obtain 20 fractions (Fr.A−Fr.T). Fraction O (0.4 g) was subjected to an ODS (50 μm) column via MPLC (MeOH/H2O, 50–100%) to give eight fractions, Fr. O1−Fr.O8. Fr. O6 (17.1 mg) was then purified with semipreparative HPLC (MeOH/H2O, 63:37, v/v; 2.0 mL/min) at 250 nm to afford 5 (6.2 mg, tR = 32.1 min). Fr. P (0.42 g) was separated with MPLC (MeOH/H2O, 60–100%) to afford five fractions, Fr. P1−Fr. P5. Fr. P4 (27.2 mg) and Fr. P5 (19.5 mg) were purified with HPLC on an RP C18 column to give 4 (7.4 mg, MeCN/H2O 40:60, 2.0 mL/min, tR = 24.9 min) and 6 (6.3 mg, MeCN/H2O 50:50, 2.0 mL/min, tR = 30.1 min), respectively. Fr. Q (0.15 g) was separated with reversed-phase ODS (50 μm) MPLC eluting with a MeOH/H2O gradient (from 60% to 100%) to afford six subfractions, Fr.Q1−Fr.Q6. Fr. Q5 (17.2 mg) was purified on an RP C18 column with HPLC (80% MeOH/H2O, 2.0 mL/min), yielding 2 (2.4 mg, tR = 28.8 min). Fr. R (1.04 g) was chromatographed over ODS via MPLC using a gradient elution of MeOH−H2O (from 50% to 100%) to get five fractions (Fr. R1−R5). Fr. R3 (475.1 mg) was then subjected to a silica gel CC (petroleum ether/EtOAc, 3:1, v:v) to give five fractions, Fr. R3a−Fr. R3e. Fr. R3c (168.0 mg) was then purified with semipreparative HPLC on an RP C18 ODS (CH3CN/H2O, 30:70, v/v; 2.0 mL/min) to afford 8 (3.5 mg, tR = 52.2 min) and 11 (116.8 mg, tR = 60.5 min). Fr. R3d (275.7 mg) was further purified with 37% CH3CN via HPLC (2.0 mL/min) to afford 7 (6.8 mg, tR = 33.0 min) and 9 (174.0 mg, tR = 39.8 min). Fr. S (0.55 g) was chromatographed over ODS using a gradient elution of MeOH/H2O (from 50% to 100%) to obtain three fractions (Fr. S1−S3). Fr. S3 (322.0 mg) was further purified with 35% CH3CN via HPLC to afford compound 10 (243.8 mg, tR = 18.6 min).
Eutypelleudesmane A (1): light-brown oil; –23.0 (c 0.10, MeOH); UV (MeOH) (log ε) λmax 241 (4.07) nm; CD (MeOH) (Δε) 242 (+17.1); IR (KBr) νmax 3357, 2956, 2929, 2873, 1741, 1650, 1455, 1438, 1376, 1232, 1153, 1116, 1068, 1024, 958, 883,850,719 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 601.4074 [M + H]+ (calcd for C36H57O7, 601.4104).
Cytosporin Y (2): light brown oil; +14.0 (c 0.10, MeOH); UV (MeOH) (log ε) λmax 241 (3.89) nm; CD (MeOH) (Δε) 238 (+9.1); IR (KBr) νmax 3359, 2956, 2927, 2857, 1454, 1376, 1261, 1014, 842, 725 cm−1; 1H and 13C NMR data, see Table 2; HRESIMS m/z 367.2125 [M + COOH]− (calcd for C20H31O6, 367.2121).
Cytosporin Z (3): light-brown oil; +12.3 (c 0.10, MeOH), +1.9 (c 0.1, CDCl3); UV (MeOH) (log ε) λmax 210 (5.37), 312 (3.13) nm; IR (KBr) νmax 3378, 2954, 2927, 2856, 1708, 1614, 1513, 1434, 1380, 1369, 1255, 1218, 1184, 1143, 1064, 1029, 977, 852 cm−1; 1H and 13C NMR data, see Table 2; HRESIMS m/z 319.1912 [M − H]− (calcd for C19H27O4, 319.1909).
Cytosporin Y1 (4): light-yellow oil; –15.8 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 221 (3.71), 240 (3.77) nm; IR νmax 3367, 2955, 2926, 2857, 1743, 1601, 1378, 1072, 1023 cm−1; CD (MeOH) (Δε) 208 (–6.4); 1H NMR and 13C NMR, see Table 3; HRESIMS m/z 363.2139 [M + Na]+ (calcd for C19H32O5Na, 363.2142).
Cytosporin Y2 (5): light-yellow oil; –60.1 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 214 (3.96), 244 (4.28) nm; IR νmax 3381, 2956, 2926, 2857, 1786, 1647, 1344, 1219, 1049, 1023, 1001, 822, 760 cm−1; CD (MeOH) (Δε) 222 (–13.2); 1H NMR and 13C NMR, see Table 3; HRESIMS m/z 389.1928 [M + Na]+ (calcd for C20H30O6Na, 389.1935).
Cytosporin Y3 (6): light-yellow oil, +37.8 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 217 (3.64), 241 (3.81) nm; IR νmax 3383, 2956, 2928, 2858, 1783, 1595, 1361, 1270, 1182, 1068, 907, 770, 737 cm−1; CD (MeOH) (Δε) 241 (+4.87); 1H NMR and 13C NMR, see Table 4; HRESIMS m/z 389.1928 [M + Na]+ (calcd for C20H30O6Na, 389.1935).
Cytosporin E1 (7): light-yellow oil, +12.9 (c 0.1, MeOH), UV (MeOH) λmax (log ε) 200 (4.10) nm; IR νmax 3393, 2926, 2856, 1783, 1464, 1361, 1184, 1158, 1086, 1058, 1023, 917, 772, 629 cm−1; CD (MeOH) (Δε) 210 (+14.2) nm; 1H NMR and 13C NMR, see Table 4; HRESIMS m/z 363.2145 407.2034 [M + Na]+ (calcd for C20H32O7Na, 407.2040).
3.4. Biological Assay
The antimicrobial activities of compounds 1–12 against Staphylococcus aureus, Escherichia coli, and Bacillus subtilis were evaluated using a previous method [16], and levofloxacin was used as a positive control. The cytotoxicities of compounds 1–12 against DU145, SW1990, Huh7, and PANC-1 human cancer cell lines were determined using the CCK-8 method [17], with cisplatin used as a positive control. The immunosuppressive activities of compounds 1–12 against ConA-induced T cell proliferation were performed as previously described [18], with cyclosporin A as a positive control.
4. Conclusions
In summary, the utilization of the OSMAC (one strain many compounds) culture strategy effectively modified the chemical profile of the Arctic-derived fungus Eutypella sp. D-1 when cultivated in different media. This approach resulted in the production of five cytosporin polyketides (compounds 1–3 and 11–12) from a rice medium and eight cytosporins (compounds 2 and 4–11) from a solid defined medium. Remarkably, compound 1 contained a unique skeleton formed by the ester linkage of two moieties: cytosporin F (12) and the eudesmane-type sesquiterpene dihydroalanto glycol. Compounds 6 and 7, characterized by a cyclic carbonate-fused cytosporin skeleton, were found to be rare in nature. However, these metabolites only exhibited weak immunosuppressive inhibitory activity against ConA-induced T cell proliferation in the antimicrobial, cytotoxic, and immunosuppressive evaluation. Collectively, this work showcased that changing the fermentation medium could be an effective strategy to trigger the production of secondary metabolites from fungi derived from the polar extreme environment.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/md21070382/s1, 1D and 2D NMR, UV, IR, and HRESMS data of 1–7.
Author Contributions
H.-B.Y. designed this study and drafted the work. H.-B.Y. and Z.N. performed the collection, extraction, isolation, and structure elucidation. Y.-P.Z. and B.H. performed the bioactive evaluation. Y.H. contributed to checking the isolation process. X.-L.L. checked the structure elucidation process. B.-H.J. and X.-Y.L. supervised the laboratory work and contributed to the critical proofreading and revision of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was financially supported by the National Key Research and Development Project (Nos. 2022YFC2804500 and 2022YFC2804105), the Natural Science Foundation of Shanghai (No.20ZR1470600), and Shanghai Pujiang Program (No.2020PJD082).
Institutional Review Board Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Arrieche, D.; Cabrera-Pardo, J.R.; San-Martin, A.; Carrasco, H.; Taborga, L. Natural Products from Chilean and Antarctic Marine Fungi and Their Biomedical Relevance. Mar. Drugs 2023, 21, 98. [Google Scholar] [CrossRef] [PubMed]
- Stevens-Miles, S.; Goetz, M.A.; Bills, G.F.; Giacobbe, R.A.; Tkacz, J.S.; Chang, R.S.; Mojena, M.; Martin, I.; Diez, M.T.; Pelaez, F. Discovery of an angiotensin II binding inhibitor from a Cytospora sp. using semi-automated screening procedures. J. Antibiot. 1996, 49, 119–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akone, S.H.; El Amrani, M.; Lin, W.; Lai, D.; Proksch, P. Cytosporins F–K, new epoxyquinols from the endophytic fungus Pestalotiopsis theae. Tetrahedron Lett. 2013, 54, 6751–6754. [Google Scholar] [CrossRef]
- Ciavatta, M.L.; Lopez-Gresa, M.P.; Gavagnin, M.; Nicoletti, R.; Manzo, E.; Mollo, E.; Guo, Y.-W.; Cimino, G. Cytosporin-related compounds from the marine-derived fungus Eutypella scoparia. Tetrahedron 2008, 64, 5365–5369. [Google Scholar] [CrossRef]
- Zhang, Y.-X.; Yu, H.-B.; Xu, W.-H.; Hu, B.; Guild, A.; Zhang, J.-P.; Lu, X.-L.; Liu, X.-Y.; Jiao, B.-H. Eutypellacytosporins A–D, Meroterpenoids from the Arctic Fungus Eutypella sp. D-1. J. Nat. Prod. 2019, 82, 3089–3095. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Müller, W.E.G.; Meier, D.; Kalscheuer, R.; Guo, Z.; Zou, K.; Umeokoli, B.O.; Liu, Z.; Proksch, P.J.M.D. Polyketide Derivatives from Mangrove Derived Endophytic Fungus Pseudopestalotiopsis theae. Mar. Drugs 2020, 18, 129–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.-B.; Wang, X.-L.; Zhang, Y.-X.; Xu, W.-H.; Zhang, J.-P.; Zhou, X.-Y.; Lu, X.-L.; Liu, X.-Y.; Jiao, B.-H. Libertellenones O–S and Eutypellenones A and B, Pimarane Diterpene Derivatives from the Arctic Fungus Eutypella sp. D-1. J. Nat. Prod. 2018, 81, 1553–1560. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.J.; Zhang, J.; Li, Y.M. Secondary metabolites from Eutypella species and their bioactivities. Mycosystema 2017, 36, 1181–1191. [Google Scholar]
- Yu, H.-B.; Wang, X.-L.; Xu, W.-H.; Zhang, Y.-X.; Qian, Y.-S.; Zhang, J.-P.; Lu, X.-L.; Liu, X.-Y. Eutypellenoids A–C, New Pimarane Diterpenes from the Arctic Fungus Eutypella sp. D-1. Mar. Drugs 2018, 16, 284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, S.; Jackson, S.A.; Patry, S.; Dobson, A.D.W. Extending the “One Strain Many Compounds” (OSMAC) Principle to Marine Microorganisms. Mar. Drugs 2018, 16, 244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, X.-D.; Zhao, N.; Xu, S.; Lü, H.-N.; Ma, S.-G.; Liu, Y.-B.; Li, Y.; Qu, J.; Yu, S.-S. Total synthesis of illicidione A and illihendione A. Tetrahedron 2015, 71, 4821–4829. [Google Scholar] [CrossRef]
- Lim, J.; Kim, I.-H.; Kim, H.H.; Ahn, K.-S.; Han, H. Enantioselective syntheses of decursinol angelate and decursin. Tetrahedron Lett. 2001, 42, 4001–4003. [Google Scholar] [CrossRef]
- Zhang, Y.-H.; Du, H.-F.; Gao, W.-B.; Li, W.; Cao, F.; Wang, C.-Y. Anti-inflammatory Polyketides from the Marine-Derived Fungus Eutypella scoparia. Mar. Drugs 2022, 20, 486. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.-X.; Sun, D.-W.; Zheng, C.-J.; Wang, C.-Y. A new hexahydrobenzopyran derivative from the gorgonian-derived Fungus Eutypella sp. Nat. Prod. Res. 2017, 31, 1640–1646. [Google Scholar] [CrossRef] [PubMed]
- Allemann, R.K. Chemical wizardry? The generation of diversity in terpenoid biosynthesis. Pure Appl. Chem. 2008, 80, 1791–1798. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.-B.; Jiao, H.; Zhu, Y.-P.; Zhang, J.-P.; Lu, X.-L.; Liu, X.-Y. Bioactive metabolites from the Arctic fungus Nectria sp. B-13. J. Asian Nat. Prod. Res. 2019, 21, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.-B.; Gu, B.-B.; Iwasaki, A.; Jiang, W.-L.; Ecker, A.; Wang, S.-P.; Yang, F.; Lin, H.-W. Dactylospenes A–E, Sesterterpenes from the Marine Sponge Dactylospongia elegans. Mar. Drugs 2020, 18, 491. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, X.; Huang, F.; Li, G.; Leadlay, P.F. Efophylins A and B, Two C2-Asymmetric Macrodiolide Immunosuppressants from Streptomyces malaysiensis. J. Nat. Prod. 2021, 84, 1579–1586. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Structures of the isolated compounds 1–12.

Figure 2.
Key COSY and HMBC correlations of 1–7.

Figure 3.
Key NOESY correlations of 1, 2, and 4–7.

Figure 4.
ECD spectra of 1, 8, and 10–12 and calculated and experimental ECD spectra of 2.

Figure 5.
Calculated and experimental ECD spectra of 4 and 5.

Figure 6.
Calculated and experimental ECD spectra of 6 and 7.

Figure 7.
Proposed biogenesis pathway of 1–12.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
1H (500 MHz) and 13C NMR (125 MHz) spectroscopic data of 1 in CDCl3.
| Position | δC | δH, mult. (J in Hz) | Position | δC | δH, mult. (J in Hz) |
|---|---|---|---|---|---|
| 2 | 76.6, C | 20 | 14.0, CH3 | 0.89, t (7.0) | |
| 3 | 73.8, CH | 3.72, m | 21 | 171.0, C | |
| 4α | 35.5, CH2 | 1.72, dd (12.5, 5.0) | 22 | 20.9, CH3 | 2.08, s |
| 4β | 2.27, t (12.0) | 23 | 37.5, CH2 | 1.35, m | |
| 5 | 55.7, C | 24 | 22.7, CH2 | 1.99, m | |
| 6 | 59.7, CH | 3.32, s | 25 | 121.2, CH | 5.31, brs |
| 7 | 64.6, CH | 4.73, s | 26 | 134.1, C | |
| 8 | 135.6, C | 27 | 46.3, CH | 1.97, m | |
| 9 | 125.1, C | 28α | 28.5, CH2 | 1.66, m | |
| 10 | 67.5, CH | 4.38, s | 28β | 1.23, m | |
| 11 | 16.0, CH3 | 1.30, s | 29 | 49.7, CH | 1.50, m |
| 12 | 27.7, CH3 | 1.29, s | 30 | 66.7, CH | 3.87, td (11.0, 5.0) |
| 13a | 61.5, CH2 | 4.67, d (12.5) | 31α | 49.4, CH2 | 1.15, m |
| 13b | 4.81, d (12.5) | 31β | 1.79, dd (12.5, 5.0) | ||
| 14 | 124.7, CH | 6.33, d (16.0) | 32 | 33.7, C | |
| 15 | 136.4, CH | 6.16, m | 33 | 39.0, CH | 1.87, m |
| 16 | 33.5, CH2 | 2.17, m | 34 | 67.5, CH2 | 3.67, dd (7.5, 4.0) |
| 17 | 28.8, CH2 | 1.42, m | 35 | 11.9, CH3 | 1.01, d (7.0) |
| 18 | 31.4, CH2 | 1.27, m | 36 | 16.5, CH3 | 0.79, s |
| 19 | 22.5, CH2 | 1.28, m | 37 | 21.2, CH3 | 1.61, s |
Table 2.
1H (500 MHz) and 13C NMR (125 MHz) spectroscopic data of 2 and 3 in CDCl3.
| 2 | 3 | ||||
|---|---|---|---|---|---|
| Position | δC | δH, mult. (J in Hz) | Position | δC | δH, mult. (J in Hz) |
| 1 | 59.3, C | 2 | 77.2, C | ||
| 2 | 57.5, CH | 3.29, s | 3 | 69.7, CH | 3.79, t (5.0) |
| 3 | 64.3, CH2 | 4.72, s | 4α | 31.5, CH2 | 3.03, dd (17.0, 5.0) |
| 4 | 131.6, C | 4β | 2.72, dd (17.0, 5.0) | ||
| 5 | 131.4, C | 5 | 118.8, C | ||
| 6 | 69.5, CH | 4.45, s | 6 | 115.1, CH | 6.61, s |
| 7 | 124.6, CH | 6.28, d (16.0) | 7 | 146.7, C | |
| 8 | 135.4, CH | 6.05, m | 8 | 123.6, C | |
| 9 | 33.5, CH2 | 2.15, m | 9 | 126.8, C | |
| 10 | 28.9, CH2 | 1.41, m | 10 | 144.8, C | |
| 11 | 31.5, CH2 | 1.28, m | 11 | 22.4, CH3 | 1.36, s |
| 12 | 22.5, CH2 | 1.29, m | 12 | 24.9, CH3 | 1.32, s |
| 13 | 14.0, CH3 | 0.88, t (6.0) | 13 | 58.8, CH2 | 4.66, s |
| 14a | 62.2, CH2 | 4.57, d (12.0) | 14 | 122.7, CH | 6.35, d (16.5) |
| 14b | 4.06, d (12.0) | 15 | 140.0, CH | 5.95, m | |
| 15α | 29.7, CH2 | 2.82, dd (15.0, 8.0) | 16 | 33.4, CH2 | 2.27, m |
| 15β | 2.30, dd (15.0, 8.0) | 17 | 29.0, CH2 | 1.50, m | |
| 16 | 117.3, CH | 5.20, t (7.0) | 18 | 31.5, CH2 | 1.35, m |
| 17 | 135.9, C | 19 | 22.6, CH2 | 1.35, m | |
| 18 | 18.0, CH3 | 1.66, s | 20 | 14.1, CH3 | 0.91, t (7.0) |
| 19 | 25.9, CH3 | 1.73, s | |||
Table 3.
1H (500 MHz) and 13C NMR (125 MHz) spectroscopic data of 4 and 5 in MeOD-d4.
| 4 | 5 | |||
|---|---|---|---|---|
| Position | δC | δH, mult. (J in Hz) | δC | δH, mult. (J in Hz) |
| 1 | 74.3, C | 84.4, C | ||
| 2 | 75.0, CH | 3.76, d (4.5) | 71.3, CH | 3.94, d (4.5) |
| 3 | 69.6, CH | 4.49, d (4.5) | 68.6, CH | 4.41, d (4.5) |
| 4 | 135.8, C | 138.3, C | ||
| 5 | 134.5, C | 127.3, C | ||
| 6 | 73.6, CH | 3.88, s | 76.9, CH | 5.15, s |
| 7 | 126.6, CH | 6.24, d (16.0) | 124.8, CH | 6.41, d (16.0) |
| 8 | 137.2, CH | 6.01, m | 136.7, CH | 6.12, dt (16.0, 7.0) |
| 9 | 35.0, CH2 | 2.17, m | 33.1, CH2 | 2.21, m |
| 10 | 30.5, CH2 | 1.46, m | 28.6, CH2 | 1.47, m |
| 11 | 32.9, CH2 | 1.28, m | 31.2, CH2 | 1.33, m |
| 12 | 23.9, CH2 | 1.34, m | 22.2, CH2 | 1.33, m |
| 13 | 14.7, CH3 | 0.91, t (7.0) | 13.0, CH3 | 0.91, t (7.0) |
| 14a | 61.1, CH2 | 4.25, d (13.0) | 58.1, CH2 | 4.13, d (13.0) |
| 14b | 4.38, d (13.0) | 4.51, d (13.0) | ||
| 15a | 35.7, CH2 | 2.60, m | 31.8, CH2 | 2.53, dd (15.0, 8.5) |
| 15b | 2.66, dd (15.0, 7.0) | |||
| 16 | 119.8, CH | 5.40, m | 115.3, CH | 5.25, m |
| 17 | 136.1, C | 137.7, C | ||
| 18 | 26.7, CH3 | 1.76, s | 24.9, CH3 | 1.77, s |
| 19 | 18.6, CH3 | 1.70, s | 16.9, CH3 | 1.68, s |
| 20 | 154.8, C | |||
Table 4.
1H (500 MHz) and 13C NMR (125 MHz) spectroscopic data of 6 and 7 in CDCl3.
| 6 | 7 | ||||
|---|---|---|---|---|---|
| Position | δC | δH, mult. (J in Hz) | Position | δC | δH, mult. (J in Hz) |
| 1 | 72.3, C | 2 | 77.7, C | ||
| 2 | 79.2, CH | 4.71, dd (8.0, 2.0) | 3 | 71.6, CH | 3.96, dd (12.0, 5.0) |
| 3 | 75.2, CH | 5.55, d (8.0) | 4α | 42.7, CH2 | 1.89, dd (12.0, 5.0) |
| 4 | 129.1, C | 4β | 2.25, d (12.0) | ||
| 5 | 139.4, C | 5 | 68.0, C | ||
| 6 | 71.5, CH | 4.14, d (2.0) | 6 | 81.3, CH | 4.66, dd (8.0, 2.0) |
| 7 | 126.1, CH | 6.52, d (16.0) | 7 | 78.5, CH | 5.23, d (8.0) |
| 8 | 136.6, CH | 6.03, dt (16.0, 7.0) | 8 | 133.3, C | |
| 9 | 34.9, CH2 | 2.21, dd (14.0, 7.0) | 9 | 135.6, C | |
| 10 | 30.4, CH2 | 1.45, m | 10 | 69.7, CH | 4.17, d (2.0) |
| 11 | 32.9, CH2 | 1.34, m | 11 | 16.9, CH3 | 1.26, s |
| 12 | 23.9, CH2 | 1.34, m | 12 | 28.5, CH3 | 1.21, s |
| 13 | 14.7, CH3 | 0.91, t (7.0) | 13a | 60.2, CH2 | 4.09, d (12.0) |
| 14a | 60.8, CH2 | 4.29, d (12.5) | 13b | 4.29, d, (12.0) | |
| 14b | 4.43, d (12.5) | 14 | 31.0, CH2 | 2.26, m | |
| 15a | 35.0, CH2 | 2.51, dd (15.0, 6.5) | 15 | 30.0, CH2 | 1.49, m |
| 15b | 2.71, dd (15.0, 8.5) | 16 | 31.1, CH2 | 2.25, m | |
| 16 | 118.8, CH | 5.38, m | 17 | 30.5, CH2 | 1.35, m |
| 17 | 136.9, C | 18 | 33.3, CH2 | 1.31, m | |
| 18 | 26.7, CH3 | 1.77, s | 19 | 24.0, CH2 | 1.32, m |
| 19 | 18.6, CH3 | 1.71, s | 20 | 14.7, CH3 | 0.9, t (7.0) |
| 20 | 156.8, C | 21 | 156.3, C | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yu, H.-B.; Ning, Z.; Hu, B.; Zhu, Y.-P.; Lu, X.-L.; He, Y.; Jiao, B.-H.; Liu, X.-Y. Cytosporin Derivatives from Arctic-Derived Fungus Eutypella sp. D-1 via the OSMAC Approach. Mar. Drugs 2023, 21, 382. https://doi.org/10.3390/md21070382
AMA Style
Yu H-B, Ning Z, Hu B, Zhu Y-P, Lu X-L, He Y, Jiao B-H, Liu X-Y. Cytosporin Derivatives from Arctic-Derived Fungus Eutypella sp. D-1 via the OSMAC Approach. Marine Drugs. 2023; 21(7):382. https://doi.org/10.3390/md21070382
Chicago/Turabian StyleYu, Hao-Bing, Zhe Ning, Bo Hu, Yu-Ping Zhu, Xiao-Ling Lu, Ying He, Bing-Hua Jiao, and Xiao-Yu Liu. 2023. "Cytosporin Derivatives from Arctic-Derived Fungus Eutypella sp. D-1 via the OSMAC Approach" Marine Drugs 21, no. 7: 382. https://doi.org/10.3390/md21070382
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.