

Untargeted Metabolomics Exploration of the Growth Stage-Dependent Chemical Space of the Sclareol-Converting Biocatalyst Hyphozyma roseonigra
 ,
,
Abstract
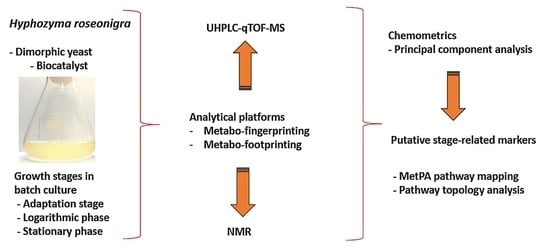
:
1. Introduction
2. Results
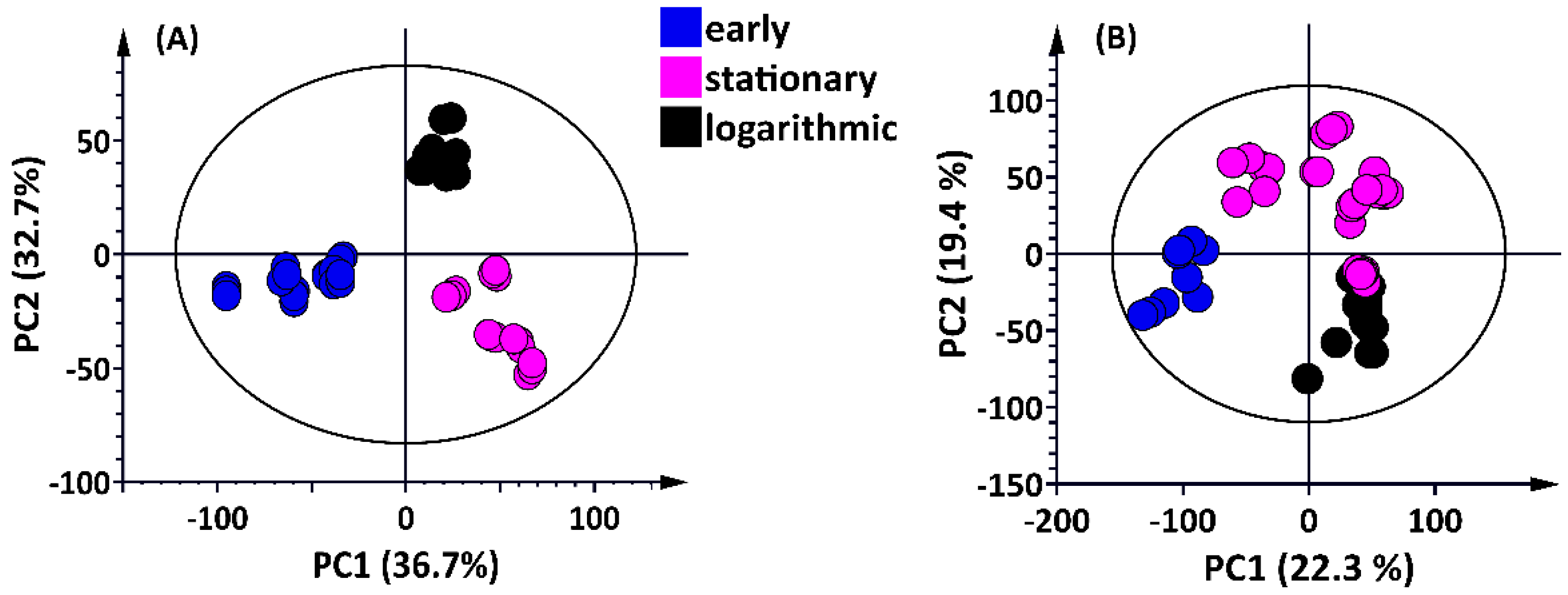
2.1. UHPLC–MS Analyses
2.1.1. Multivariate Data Analysis
2.1.2. Metabolite Annotation and Relative Quantification of UHPLC–MS Analyzed Compounds
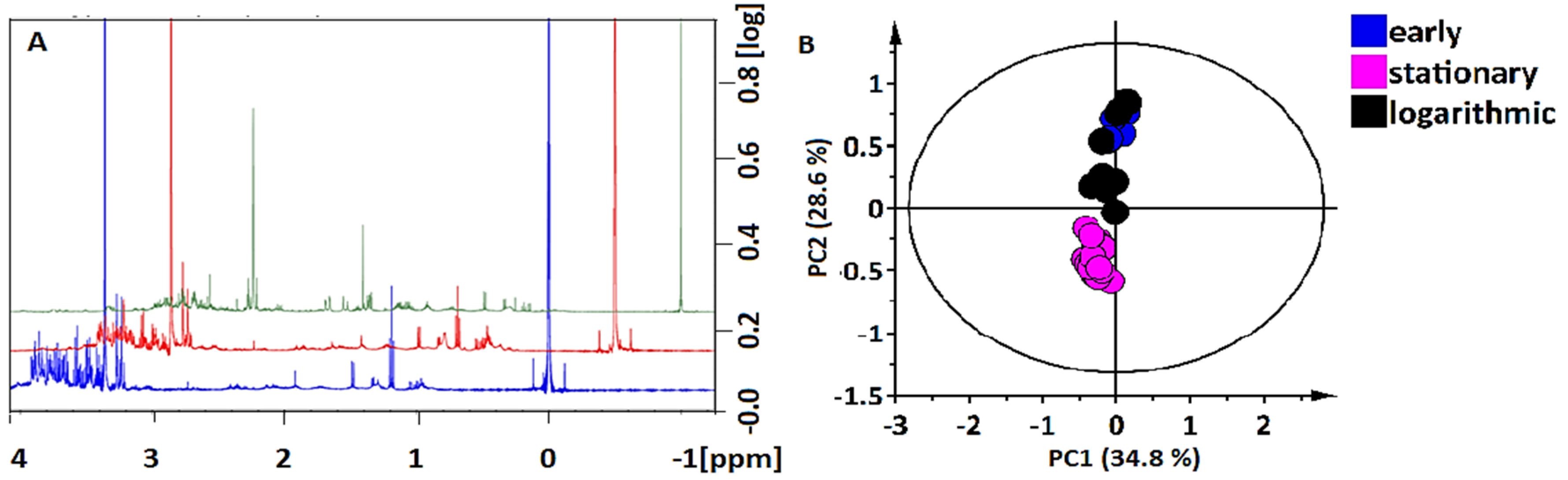
2.2. NMR Multivariate Analyses, Metabolite Annotation and Relative Quantification
Metabolite Annotation and Relative Quantification of NMR Analyzed Compounds
2.3. Pathway Mapping and Network Correlation Analysis
3. Discussion
3.1. UHPLC–MS and Multivariate Data Analyses
3.2. NMR and Multivariate Analyses
3.3. Pathway Mapping and Network Correlation Analysis
4. Materials and Methods
4.1. Growth of Hyphozyma roseonigra
4.2. Harvesting of Cells, Metabolite Extraction, and Sample Preparation
4.3. Ultra-High Performance Liquid Chromatography–Mass Spectrometry (UHPLC–MS)
4.4. Nuclear Magnetic Resonance Analysis
4.5. Data Processing for LC–MS and NMR
4.6. Multivariate Data Analysis and Statistical Modeling
4.7. Metabolite Annotation
4.8. Metabolomics Pathway Analysis and Network Correlation Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wohlgemuth, R. Biocatalysis—Key to sustainable industrial chemistry. Curr. Opin. Biotechnol. 2010, 21, 713–724. [Google Scholar] [CrossRef]
- Allendes, J.A.; Bustos, D.A.; Pacciaroni, A.D.V.; Sosa, V.E.; Bustos, D.A. Microbial functionalization of (–)-ambroxide by filamentous fungi. Biocatal. Biotransform. 2011, 29, 83–86. [Google Scholar] [CrossRef]
- Cheetham, P.S.J. The use of biotransformations for the production of flavours and fragrances. Trends Biotechnol. 1993, 11, 478–488. [Google Scholar] [CrossRef]
- De Hoog, G.S.; Smith, M.T. Key to the species of Hyphozyma (yeast-like Hyphomycetes) and description of H. roseonigra sp. nov. Antonie van Leeuwenhoek 1986, 52, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Farbood, M.I.; Willis, B.J. Process for Producing Diol and Furan and Microorganism Capable of Same. U.S. Patent 4798799A, 17 January 1989. [Google Scholar]
- Steenkamp, L.H.; Taka, M. Process for the Production of Ambrafuran. U.S. Patent 2010/0248316 A1, 30 September 2010. [Google Scholar]
- Ncube, E.N.; Steenkamp, L.H.; Dubery, I.A. Ambrafuran (AmbroxTM) synthesis from natural plant product precursors. Molecules 2020, 25, 3851. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Rochfort, S. Recent progress in polar metabolite quantification in plants using liquid chromatography—Mass spectrometry (LC-MS). J. Integr. Plant Biol. 2014, 56, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Theodoridis, G.A.; Gika, H.G.; Plumb, R.; Wilson, I.D. Liquid chromatographic methods combined with mass spectrometry in metabolomics. In Proteomic and Metabolomic Approaches to Biomarker Discovery; Issaq, H., Veenstra, T., Eds.; Elsevier Inc.: Amsterdam, NL, USA, 2020; pp. 149–169. [Google Scholar] [CrossRef]
- Şenyuva, H.Z.; Gilbert, J.; Öztürkoǧlu, S. Rapid analysis of fungal cultures and dried figs for secondary metabolites by LC/TOF-MS. Anal. Chim. Acta 2008, 617, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Porzel, A.; Farag, M.A.; Mülbradt, J.; Wessjohann, L.A. Metabolite profiling and fingerprinting of Hypericum species: A comparison of MS and NMR metabolomics. Metabolomics 2014, 10, 574–588. [Google Scholar] [CrossRef]
- Forgue, P.; Halouska, S.; Werth, M.; Xu, K.; Harris, S.; Powers, R. NMR metabolic profiling of Aspergillus nidulans to monitor drug and protein activity. J. Proteome Res. 2006, 5, 1916–1923. [Google Scholar] [CrossRef]
- Ząbek, A.; Klimek-Ochab, M.; Jawień, E.; Młynarz, P. Biodiversity in targeted metabolomics analysis of filamentous fungal pathogens by 1H NMR-based studies. World J. Microbiol. Biotechnol. 2017, 33, 132. [Google Scholar] [CrossRef] [Green Version]
- Moco, S.; Bino, R.J.; De Vos, R.C.H.; Vervoort, J. Metabolomics technologies and metabolite identification. Trends Anal. Chem. 2007, 26, 55–866. [Google Scholar] [CrossRef]
- Diao, M.; Li, C.; Li, J.; Lu, J.; Xie, N. Probing the biotransformation process of sclareol by resting cells of Hyphozyma roseonigra. J. Agric. Food Chem. 2022, 70, 10563–10570. [Google Scholar] [CrossRef] [PubMed]
- Ncube, E.N.; Steenkamp, L.H.; Dubery, I.A. GC–MS based profiling of alkanes in the filamentous yeast Hyphozyma roseonigra (Moesziomyces antarcticus). Biotechnol. Lett. 2019, 41, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Ncube, E.N.; Steenkamp, P.A.; van der Westhuyzen, C.; Steenkamp, L.H.; Dubery, I.A. Metabolomics-guided analysis of the biocatalytic conversion of sclareol to ambradiol by Hyphozyma roseonigra. Catalysts 2022, 12, 55. [Google Scholar] [CrossRef]
- Ncube, E.N.; Mathiba, K.; Steenkamp, L.H.; Dubery, I.A. Gas chromatographic profiling of the biocatalytic conversion of sclareol to ambradiol by Hyphozyma roseonigra. Biocatal. Biotransform. 2022, 40, 308–312. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, X.; Yao, Q.; Hu, D.; Qin, H. Comparative proteomic analyses of Hyphozyma roseonigra ATCC 20624 in response to sclareol. Braz. J. Microbiol. 2019, 50, 79–84. [Google Scholar] [CrossRef] [Green Version]
- Carlson, R.; Tugizimana, F.; Steenkamp, P.A.; Dubery, I.A.; Labuschagne, N. Differential metabolic reprogramming in Paenibacillus alvei-primed Sorghum bicolor seedlings in response to Fusarium pseudograminearum infection. Metabolites 2019, 9, 150. [Google Scholar] [CrossRef] [Green Version]
- Xia, J.; Wishart, D.S. Metabolomic data processing, analysis, and interpretation using MetaboAnalyst. Curr. Protoc. Bioinformatics 2011, 34, 14.10.1–14.10.48. [Google Scholar] [CrossRef]
- Fukushima, A.; Kusano, M. Recent progress in the development of metabolome databases for plant systems biology. Front. Plant Sci. 2013, 4, 73. [Google Scholar] [CrossRef]
- De Vijlder, T.; Valkenborg, D.; Lemière, F.; Romijn, E.P.; Laukens, K.; Cuyckens, F. A tutorial in small molecule identification via electrospray ionization-mass spectrometry: The practical art of structural elucidation. Mass Spectrom. Rev. 2018, 37, 607–629. [Google Scholar] [CrossRef]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, H.M.; Beger, R.; Daykin, C.A.; Fan, T.W.M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis: Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostidis, S.; Addie, R.D.; Morreau, H.; Mayboroda, O.A.; Giera, M. Quantitative NMR analysis of intra- and extracellular metabolism of mammalian cells: A tutorial. Anal. Chim. Acta 2017, 980, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Marante Toledo, F.J.; Mioso, R.; Barrera, J.B.; González González, J.E.; Santana Rodríguez, J.J.; De Laguna, I.H.B. Structural characterization and metabolite profiling of the facultative marine fungus Paecilomyces variotii. Ann. Microbiol. 2012, 6, 1601–1607. [Google Scholar] [CrossRef]
- Marshall, D.D.; Powers, R. Beyond the paradigm: Combining mass spectrometry and nuclear magnetic resonance for metabolomics. Prog. Nucl. Magn. Reson. Spectrosc. 2017, 100, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Wittmann, C.; Hans, M.; van Winden, W.A.; Ras, C.; Heijnen, J.J. Dynamics of intracellular metabolites of glycolysis and TCA cycle during cell-cycle-related oscillation in Saccharomyces cerevisiae. Biotechnol. Bioeng. 2005, 89, 839–847. [Google Scholar] [CrossRef]
- Jewett, M.C.; Hofmann, G.; Nielsen, J. Fungal metabolite analysis in genomics and phenomics. Curr. Opin. Biotechnol. 2006, 17, 191–197. [Google Scholar] [CrossRef]
- Calvo, A.M.; Wilson, R.A.; Bok, J.W.; Keller, N.P. Relationship between secondary metabolism and fungal development. Microbiol. Mol. Biol. Rev. 2002, 66, 447–459. [Google Scholar] [CrossRef] [Green Version]
- Aittokallio, T.; Schwikowski, B. Graph-based methods for analysing networks in cell biology. Brief Bioinfor. 2006, 7, 243–255. [Google Scholar] [CrossRef]
- Xia, J.; Wishart, D.S. MetPA: A web-based metabolomics tool for pathway analysis and visualization. Bioinformatics 2010, 26, 2342–2344. [Google Scholar] [CrossRef]
- Pócsi, I.; Prade, R.A.; Penninckx, M.J. Glutathione, altruistic metabolite in fungi. Adv. Microb. Physiol. 2004, 49, 1–76. [Google Scholar] [CrossRef]
- Ratledge, C.; Wynn, J.P. The biochemistry and molecular biology of lipid accumulation in oleaginous microorganisms. Adv. Appl. Microbiol. 2002, 51, 1–52. [Google Scholar] [CrossRef] [PubMed]
- Herman, N.A.; Zhang, W. Enzymes for fatty acid-based hydrocarbon biosynthesis. Curr. Opin. Chem. Biol. 2016, 35, 22–28. [Google Scholar] [CrossRef]
- Tang, X.; Lee, J.; Ning, W. Engineering the fatty acid metabolic pathway in Saccharomyces cerevisiae for advanced biofuel production. Metab. Eng. Commun. 2015, 2, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Breuninger, M.; Trujillo, C.G.; Serrano, E.; Fischer, R.; Requena, N. Different nitrogen sources modulate activity but not expression of glutamine synthetase in arbuscular mycorrhizal fungi. Fungal Genet. Biol. 2004, 41, 542–552. [Google Scholar] [CrossRef]
- Jeffries, T.W. Utilization of xylose by bacteria, yeasts, and fungi. Adv. Biochem. Eng. Biotechnol. 1983, 27, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Kohlhaw, G.B. Leucine biosynthesis in fungi: Entering metabolism through the back door. Microbiol. Mol. Biol. Rev. 2003, 67, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Cossins, E.A.; Chen, L. Folates and one-carbon metabolism in plants and fungi. Phytochemistry 1997, 45, 437–452. [Google Scholar] [CrossRef]
- Estramareix, B.; Thérisod, M. Biosynthesis of thiamine: 5-aminoimidazole ribotide as the precursor of all the carbon atoms of the pyrimidine moiety. J. Am. Chem. Soc. 1984, 106, 3857–3860. [Google Scholar] [CrossRef]
- Eisenreich, W.; Bacher, A.; Arigoni, D.; Rohdich, F. Biosynthesis of isoprenoids via the non-mevalonate pathway. Cell. Mol. Life Sci. 2004, 61, 1401–1426. [Google Scholar] [CrossRef]
- Lim, F.Y.; Sanchez, J.F.; Wang, C.C.C.; Keller, N.P. Toward awakening cryptic secondary metabolite gene clusters in filamentous fungi. Meth. Enzymol. 2012, 517, 303–324. [Google Scholar] [CrossRef] [Green Version]
- Caniard, A.; Zerbe, P.; Legrand, S.; Cohade, A.; Valot, N.; Magnard, J.-L.; Bohlmann, J.; Legendre, L. Discovery and functional characterization of two diterpene synthases for sclareol biosynthesis in Salvia sclarea (L.) and their relevance for perfume manufacture. BMC Plant Biol. 2012, 12, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Guido, S.I.; Sengupta, D.; Nápoles-Rivera, F.; González-Campos, J.B.; Del Río, R.B.; Ponce-Ortega, J.M.; El-Halwagi, M.M. Life cycle assessment for Ambrox® production from different chemical routes. J. Clean Prod. 2016, 130, 202–212. [Google Scholar] [CrossRef]
- Dettmer, K.; Aronov, P.A.; Hammock, B.D. Mass spectrometry-based metabolomics, Mass Spectrom. Rev. 2007, 26, 51–78. [Google Scholar] [CrossRef]
- Saccenti, E.; Hoefsloot, H.C.J.; Smilde, A.K.; Westerhuis, J.A.; Hendriks, M.M.W.B. Reflections on univariate and multivariate analysis of metabolomics data. Metabolomics 2014, 10, 361–374. [Google Scholar] [CrossRef]
- Tugizimana, F.; Piater, L.A.; Dubery, I.A. Plant metabolomics: A new frontier in phytochemical analysis. S. Afr. J. Sci. 2013, 109, 18–20. [Google Scholar] [CrossRef] [Green Version]
- Wiklund, S.; Johansson, E.; Sjöström, L.; Mellerowicz, E.J.; Edlund, U.; Shockcor, J.P.; Gottfries, J.; Moritz, T.; Trygg, J. Visualization of GC/TOF-MS-based metabolomics data for identification of biochemically interesting compounds using OPLS class models. Anal. Chem. 2008, 80, 115–122. [Google Scholar] [CrossRef]
- Trivedi, K.D.; Iles, K.R. The application of SIMCA P+ in shotgun metabolomics analysis of ZIC®HILIC-MS spectra of human urine—Experience with the Shimadzu IT-TOF and profiling solutions data extraction software. J. Chromatogr. Sep. Tech. 2012, 3, 145. [Google Scholar] [CrossRef] [Green Version]
- Airoldi, C.; Tripodi, F.; Guzzi, C.; Nicastroab, R.; Coccetti, P. NMR analysis of budding yeast metabolomics: A rapid method for sample preparation. Mol. Biosyst. 2015, 11, 379–383. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assigned Feature Number | Metabolite Annotation | Biological Role | Intra- Cellular | Extra- Cellular |
|---|---|---|---|---|
| Acetone extracts | ||||
| 1 | 5-Aminoimidazole ribonucleotide | Intermediate | ✓ | |
| 2 | Pantothenic acid | Vitamin | ✓ | |
| 3 | Thymine diphosphate | Nucleotide | ✓ | |
| 4 | Glycyl-leucine | Dipeptide | ✓ | |
| 5 | Xanthosine-5-phosphate | Purine | ✓ | |
| 6 | N6,N6-Dimethyladenosine | Ribonucleoside | ✓ | |
| 7 | Maculosin | Dipeptide | ✓ | |
| 8 | 2-Deoxyribose-1-phosphate | Intermediate | ✓ | |
| 9 | S-Adenosyl-L-homo-cysteine | Methyl donor | ✓ | |
| 10 | 18-Acetoxy-1-alpha-25-dihydroxy vit. D3 | Vitamin | ✓ | |
| 11 | 5-Methyl-tetrahydrofolate | DNA synthesis | ✓ | |
| 12 | Reynosin | Diterpene | ✓ | |
| 13 | Broussonin C | Polyketide/phenolic | ✓ | |
| Methanol extracts | ||||
| 14 | Deoxyuridine | Ribonucleoside | ✓ | |
| 15 | Biotin | Vitamin | ✓ | |
| 16 | UDP-glucuronate | Sugar | ✓ | |
| 17 | Xanthosine-5-phosphate | Purine | ✓ | |
| Assigned Feature Number | Metabolite Annotation | Assigned Feature Number | Metabolite Annotation |
|---|---|---|---|
| Amino acids and derivatives | |||
| 18 | Aspartate | 19 | Glutamine |
| 20 | Valine | 21 | Threonine |
| 22 | Alanine | 23 | Glutamate |
| 24 | Isoleucine | 25 | Leucine |
| 26 | Cysteine | 27 | Ornithine |
| 28 | Serine | 29 | N-acetyl aspartate |
| Fatty acids and derivatives | |||
| 30 | Linoleic acid | 31 | Hexadecyl octanoate ester |
| 32 | Nonadecanoic acid | 33 | Palmitic acid (OCH3) |
| 34 | 2-Hydroxy-isobutyric acid | ||
| Sugars and derivatives | |||
| 35 | Galactose-1-phosphate | 36 | Glucose |
| 37 | Galactitol | 38 | Myo-inositol |
| Organic acids | |||
| 39 | Lactate | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ncube, E.N.; Sitole, L.; Steenkamp, P.A.; Steenkamp, L.H.; Dubery, I.A. Untargeted Metabolomics Exploration of the Growth Stage-Dependent Chemical Space of the Sclareol-Converting Biocatalyst Hyphozyma roseonigra. Catalysts 2022, 12, 1225. https://doi.org/10.3390/catal12101225
Ncube EN, Sitole L, Steenkamp PA, Steenkamp LH, Dubery IA. Untargeted Metabolomics Exploration of the Growth Stage-Dependent Chemical Space of the Sclareol-Converting Biocatalyst Hyphozyma roseonigra. Catalysts. 2022; 12(10):1225. https://doi.org/10.3390/catal12101225
Chicago/Turabian StyleNcube, Efficient N., Lungile Sitole, Paul A. Steenkamp, Lucia H. Steenkamp, and Ian A. Dubery. 2022. "Untargeted Metabolomics Exploration of the Growth Stage-Dependent Chemical Space of the Sclareol-Converting Biocatalyst Hyphozyma roseonigra" Catalysts 12, no. 10: 1225. https://doi.org/10.3390/catal12101225