Impact of Physicochemical Parameters on the Diversity and Distribution of Microbial Communities Associated with Three South African Peatlands


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Determination of Physicochemical Properties
2.3. DNA Extraction and Library Preparation
2.4. Data Processing
2.4.1. Metataxonomics
2.4.2. Community Analyses
3. Results
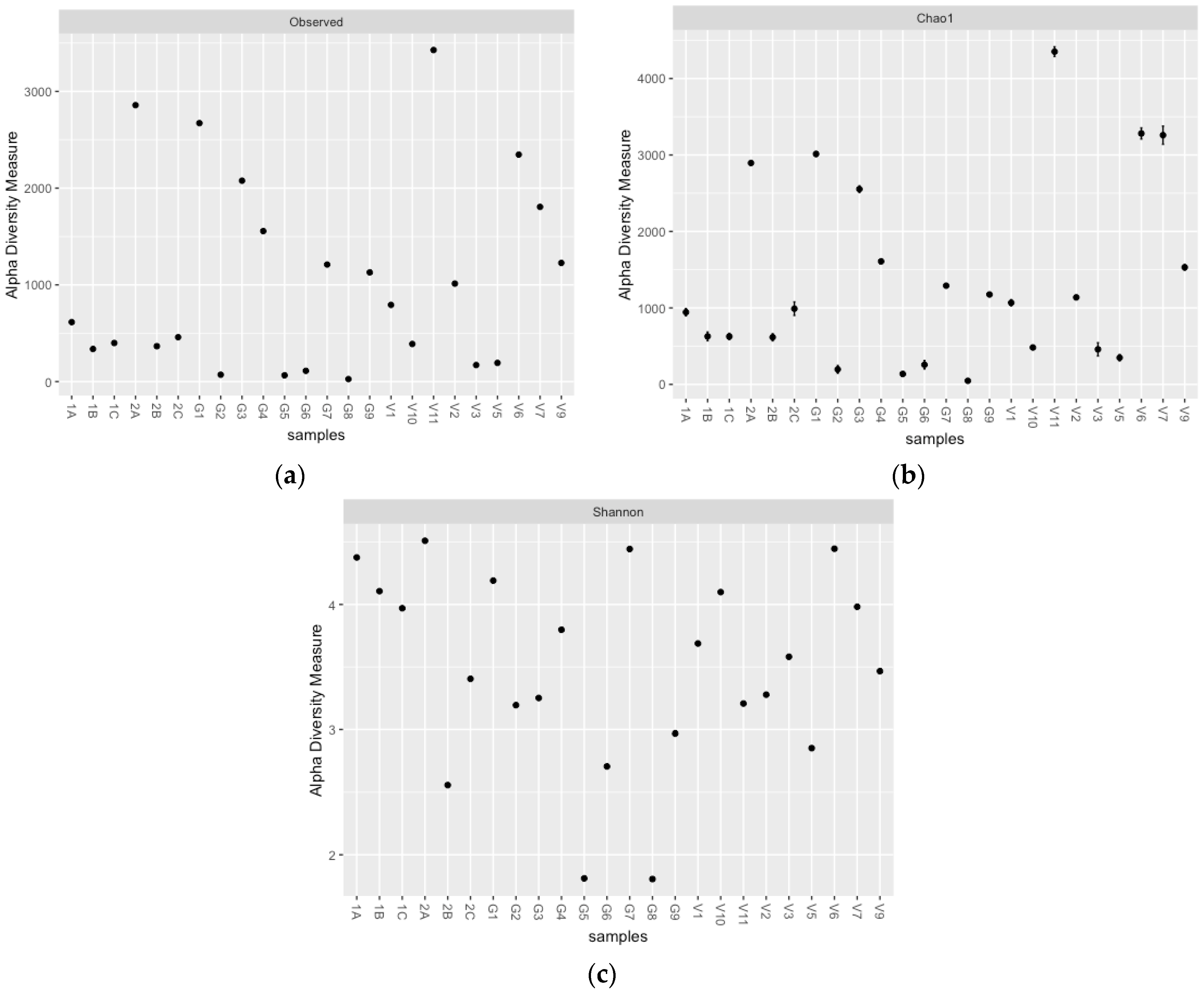
3.1. Actinobacterial and Fungal Community Structures at Different Sites and Depths
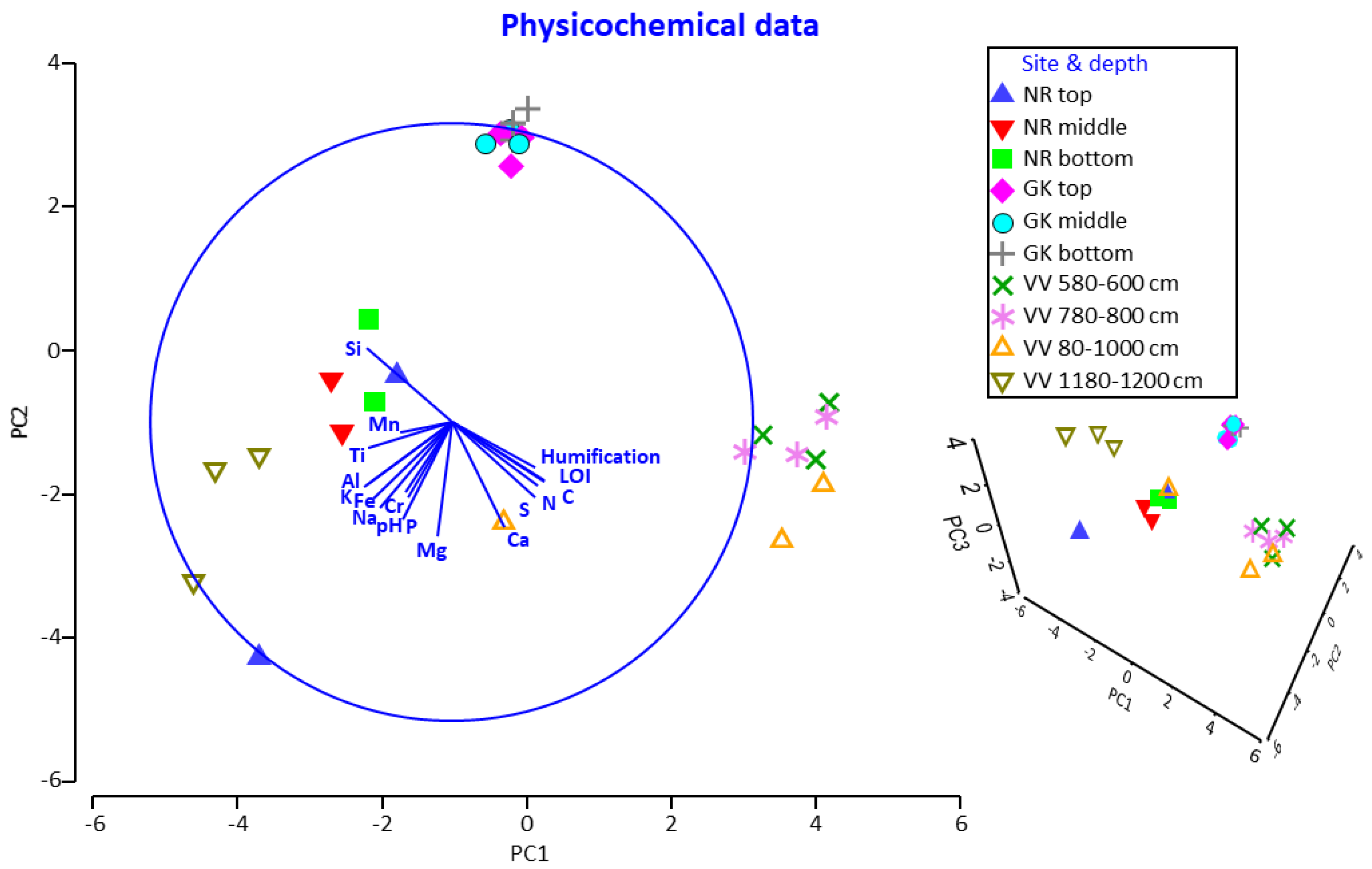
3.2. Physicochemical Profiles at Different Peatland Sites and Depths
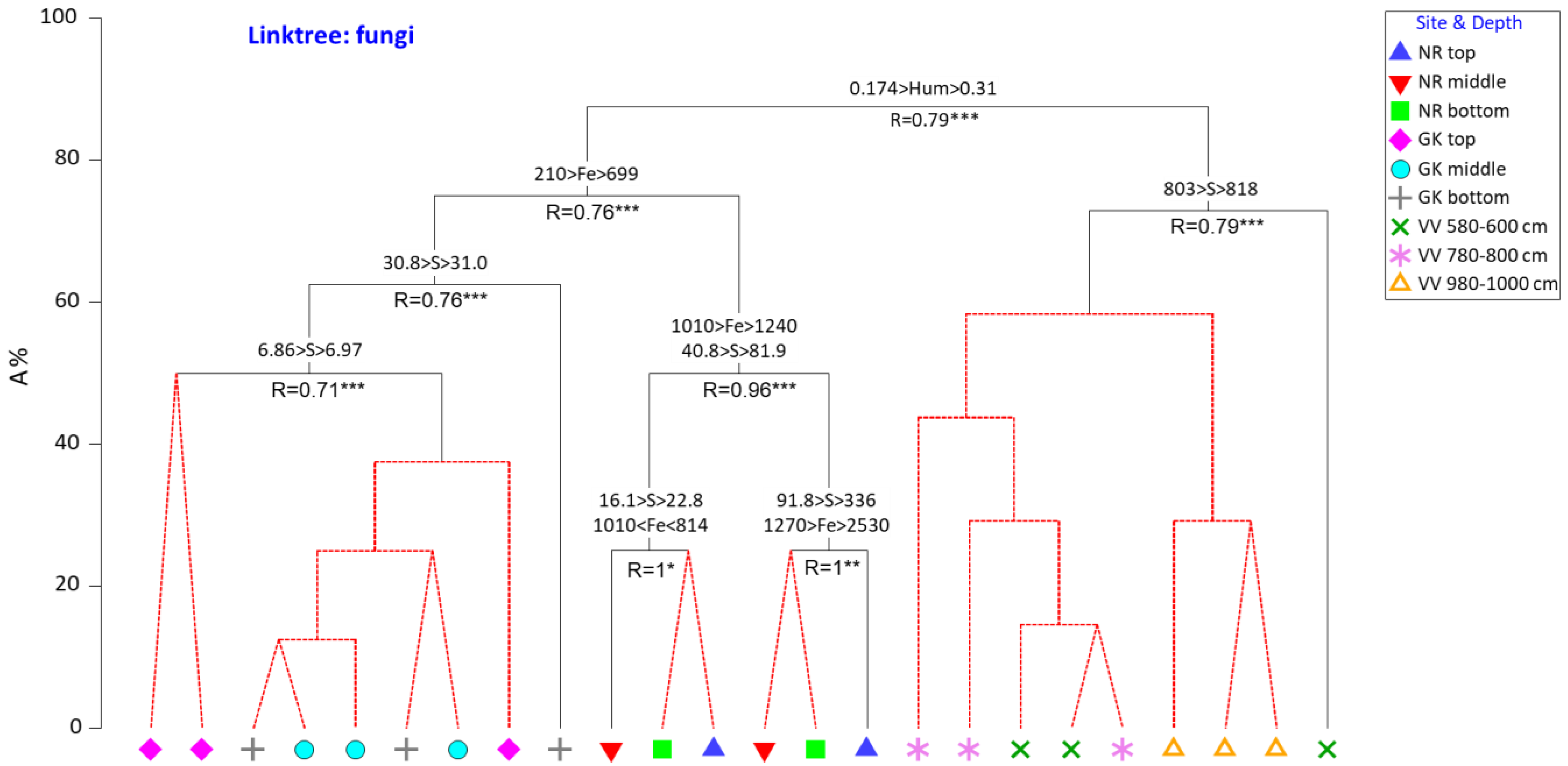
3.3. Physicochemical Parameters as Drivers for Actinobacterial and Fungal Community Compositions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ivanova, A.A.; Beletsky, A.V.; Rakitin, A.L.; Kadnikov, V.V.; Philippov, D.A.; Mardanov, A.V.; Ravin, N.V.; Dedysh, S.N. Closely Located but Totally Distinct: Highly Contrasting Prokaryotic Diversity Patterns in Raised Bogs and Eutrophic Fens. Microorganisms 2020, 8, 484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellery, W.N.; Grenfell, S.E.; Grenfell, M.C.; Humphries, M.S.; Barnes, K.; Dahlberg, A.; Kindness, A. Peat Formation in the Context of the Development of the Mkuze Floodplain on the Coastal Plain of Maputaland, South Africa. Geomorphology 2012, 141–142, 11–20. [Google Scholar] [CrossRef]
- Kusai, N.A.; Ayob, Z.; Maidin, M.S.T.; Safari, S.; Ahmad Ali, S.R. Characterization of Fungi from Different Ecosystems of Tropical Peat in Sarawak, Malaysia. Rend. Fis. Acc. Lincei 2018, 29, 469–482. [Google Scholar] [CrossRef]
- Liu, B.; Talukder, M.J.H.; Terhonen, E.; Lampela, M.; Vasander, H.; Sun, H.; Asiegbu, F. The Microbial Diversity and Structure in Peatland Forest in Indonesia. Soil Use Manag. 2020, 36, 123–138. [Google Scholar] [CrossRef]
- Tfaily, M.M.; Hodgkins, S.; Podgorski, D.C.; Chanton, J.P.; Cooper, W.T. Comparison of Dialysis and Solid-Phase Extraction for Isolation and Concentration of Dissolved Organic Matter Prior to Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal. Bioanal. Chem. 2012, 404, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Andersen, R.; Chapman, S.J.; Artz, R.R.E. Microbial Communities in Natural and Disturbed Peatlands: A Review. Soil Biol. Biochem. 2013, 57, 979–994. [Google Scholar] [CrossRef]
- Girkin, N.T.; Lopes dos Santos, R.A.; Vane, C.H.; Ostle, N.; Turner, B.L.; Sjögersten, S. Peat Properties, Dominant Vegetation Type and Microbial Community Structure in a Tropical Peatland. Wetlands 2020, 40, 1367–1377. [Google Scholar] [CrossRef] [Green Version]
- Xue, D.; Liu, T.; Chen, H.; Liu, J.; Hu, J.; Liu, L. Fungi Are More Sensitive than Bacteria to Drainage in the Peatlands of the Zoige Plateau. Ecol. Indic. 2021, 124, 107367. [Google Scholar] [CrossRef]
- Dom, S.P.; Ikenaga, M.; Lau, S.Y.L.; Radu, S.; Midot, F.; Yap, M.L.; Chin, M.-Y.; Lo, M.L.; Jee, M.S.; Maie, N.; et al. Linking Prokaryotic Community Composition to Carbon Biogeochemical Cycling across a Tropical Peat Dome in Sarawak, Malaysia. Sci. Rep. 2021, 11, 6416. [Google Scholar] [CrossRef]
- Gründling, P.-L.; Grobler, R. Peatlands and Mires of South Africa. Stapfia 2005, 85, 379–396. [Google Scholar]
- Andersen, R.; Rochefort, L.; Poulin, M. Peat, Water and Plant Tissue Chemistry Monitoring: A Seven-Year Case-Study in a Restored Peatland. Wetlands 2010, 30, 159–170. [Google Scholar] [CrossRef]
- Andersen, R.; Wells, C.; Macrae, M.; Price, J. Nutrient Mineralisation and Microbial Functional Diversity in a Restored Bog Approach Natural Conditions 10 Years Post Restoration. Soil Biol. Biochem. 2013, 64, 37–47. [Google Scholar] [CrossRef]
- Nwaishi, F.; Petrone, R.M.; Price, J.S.; Ketcheson, S.J.; Slawson, R.; Andersen, R. Impacts of Donor-Peat Management Practices on the Functional Characteristics of a Constructed Fen. Ecol. Eng. 2015, 81, 471–480. [Google Scholar] [CrossRef]
- Grundling, P.-L. The Quaternary Peat Deposits of Maputaland, Northern Kwazulu-Natal, South Africa: Categorisation, Chronology and Utilisation. 2001—ProQuest. Available online: https://www.proquest.com/openview/dac25383bdeb3495a3765e9a6ade825c/1?pq-origsite=gscholar&cbl=2026366&diss=y (accessed on 16 August 2022).
- Strobel, P.; Kasper, T.; Frenzel, P.; Schittek, K.; Quick, L.J.; Meadows, M.E.; Mäusbacher, R.; Haberzettl, T. Late Quaternary Palaeoenvironmental Change in the Year-Round Rainfall Zone of South Africa Derived from Peat Sediments from Vankervelsvlei. Quat. Sci. Rev. 2019, 218, 200–214. [Google Scholar] [CrossRef]
- Chambers, F.M.; Beilman, D.W.; Yu, Z. Methods for Determining Peat Humification and for Quantifying Peat Bulk Density, Organic Matter and Carbon Content for Palaeostudies of Climate and Peatland Carbon Dynamics. Mires Peats 2011, 7, 1–10. [Google Scholar]
- Blackford, J.J.; Chambers, F.M. Determining the Degree of Peat Decomposition for Peat-Based Paleoclimatic Studies. Int. Peat J. (Finland) 1993, 8, 5. [Google Scholar]
- Schäfer, J.; Jäckel, U.; Kämpfer, P. Development of a New PCR Primer System for Selective Amplification of Actinobacteria. FEMS Microbiol. Lett. 2010, 311, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Gardes, M.; Bruns, T.D. ITS Primers with Enhanced Specificity for Basidiomycetes - Application to the Identification of Mycorrhizae and Rusts. Mol. Ecol. 1993, 2, 113–118. [Google Scholar] [CrossRef] [PubMed]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J.W. Amplification and Direct Sequencing of Fungal Ribosomal RNA Genes for Phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press Inc.: New York, NY, USA, 1990; pp. 315–322. [Google Scholar]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME Allows Analysis of High-Throughput Community Sequencing Data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.-H.; Ha, S.-M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A Taxonomically United Database of 16S RRNA Gene Sequences and Whole-Genome Assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef]
- Kõljalg, U.; Nilsson, H.R.; Schigel, D.; Tedersoo, L.; Larsson, K.-H.; May, T.W.; Taylor, A.F.S.; Jeppesen, T.S.; Frøslev, T.G.; Lindahl, B.D.; et al. The Taxon Hypothesis Paradigm—On the Unambiguous Detection and Communication of Taxa. Microorganisms 2020, 8, 1910. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.H.; Song, Z.; Bates, S.T.; Branco, S.; Tedersoo, L.; Menke, J.; Schilling, J.S.; Kennedy, P.G. FUNGuild: An Open Annotation Tool for Parsing Fungal Community Datasets by Ecological Guild. Fungal Ecol. 2016, 20, 241–248. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Clarke, K.R.; Gorley, R.N. Getting Started with PRIMER V7; PRIMER-E: Plymouth, UK, 2015; Volume 20. [Google Scholar]
- Clarke, K.R.; Somerfield, P.J.; Gorley, R.N. Clustering in Non-Parametric Multivariate Analyses. J. Exp. Mar. Biol. Ecol. 2016, 483, 147–155. [Google Scholar] [CrossRef] [Green Version]
- Briones, M.J.I.; Juan-Ovejero, R.; McNamara, N.P.; Ostle, N.J. Microbial “Hotspots” of Organic Matter Decomposition in Temperate Peatlands Are Driven by Local Spatial Heterogeneity in Abiotic Conditions and Not by Vegetation Structure. Soil Biol. Biochem. 2022, 165, 108501. [Google Scholar] [CrossRef]
- Lew, S.; Glińska-Lewczuk, K.; Ziembińska-Buczyńska, A. Prokaryotic Community Composition Affected by Seasonal Changes in Physicochemical Properties of Water in Peat Bog Lakes. Water 2018, 10, 485. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Tian, J.; Bu, Z.; Lamit, L.J.; Chen, H.; Zhu, Q.; Peng, C. Structural and Functional Differentiation of the Microbial Community in the Surface and Subsurface Peat of Two Minerotrophic Fens in China. Plant Soil 2019, 437, 21–40. [Google Scholar] [CrossRef]
- Aksenov, A.S.; Shirokova, L.S.; Kisil, O.Y.; Kolesova, S.N.; Lim, A.G.; Kuzmina, D.; Pouillé, S.; Alexis, M.A.; Castrec-Rouelle, M.; Loiko, S.V.; et al. Bacterial Number and Genetic Diversity in a Permafrost Peatland (Western Siberia): Testing a Link with Organic Matter Quality and Elementary Composition of a Peat Soil Profile. Diversity 2021, 13, 328. [Google Scholar] [CrossRef]
- Kotiaho, M.; Fritze, H.; Merilä, P.; Tuomivirta, T.; Väliranta, M.; Korhola, A.; Karofeld, E.; Tuittila, E.-S. Actinobacteria Community Structure in the Peat Profile of Boreal Bogs Follows a Variation in the Microtopographical Gradient Similar to Vegetation. Plant Soil 2013, 369, 103–114. [Google Scholar] [CrossRef]
- Hao, J.; Chai, Y.N.; Lopes, L.D.; Ordóñez, R.A.; Wright, E.E.; Archontoulis, S.; Schachtman, D.P. The Effects of Soil Depth on the Structure of Microbial Communities in Agricultural Soils in Iowa (United States). Appl. Environ. Microbiol. 2021, 87, e02673-20. [Google Scholar] [CrossRef]
- Jenkins, S.N.; Waite, I.S.; Blackburn, A.; Husband, R.; Rushton, S.P.; Manning, D.C.; O’Donnell, A.G. Actinobacterial Community Dynamics in Long Term Managed Grasslands. Antonie Leeuwenhoek 2009, 95, 319–334. [Google Scholar] [CrossRef] [PubMed]
- Oloo, F.; Valverde, A.; Quiroga, M.V.; Vikram, S.; Cowan, D.; Mataloni, G. Habitat Heterogeneity and Connectivity Shape Microbial Communities in South American Peatlands. Sci. Rep. 2016, 6, 25712. [Google Scholar] [CrossRef] [Green Version]
- McHugh, T.A.; Schwartz, E. A Watering Manipulation in a Semiarid Grassland Induced Changes in Fungal but Not Bacterial Community Composition. Pedobiologia 2016, 59, 121–127. [Google Scholar] [CrossRef]
- Moll, J.; Klingenfuss, F.; Widmer, F.; Gogos, A.; Bucheli, T.D.; Hartmann, M.; van der Heijden, M.G.A. Effects of Titanium Dioxide Nanoparticles on Soil Microbial Communities and Wheat Biomass. Soil Biol. Biochem. 2017, 111, 85–93. [Google Scholar] [CrossRef]
- Schlatter, D.C.; Reardon, C.L.; Johnson-Maynard, J.; Brooks, E.; Kahl, K.; Norby, J.; Huggins, D.; Paulitz, T.C. Mining the Drilosphere: Bacterial Communities and Denitrifier Abundance in a No-Till Wheat Cropping System. Front. Microbiol. 2019, 10, 1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tkhakakhova, A.K.; Chernov, T.I.; Ivanova, E.A.; Kutovaya, O.V.; Kogut, B.M.; Zavalin, A.A. Changes in the Metagenome of the Prokaryotic Community of Chernozems under the Influence of Mineral Fertilizers. Russ. Agric. Sci. 2016, 42, 62–65. [Google Scholar] [CrossRef]
- Pershina, E.V.; Ivanova, E.A.; Korvigo, I.O.; Chirak, E.L.; Sergaliev, N.H.; Abakumov, E.V.; Provorov, N.A.; Andronov, E.E. Investigation of the Core Microbiome in Main Soil Types from the East European Plain. Sci. Total Environ. 2018, 631–632, 1421–1430. [Google Scholar] [CrossRef]
- Zhang, B.; Wu, X.; Tai, X.; Sun, L.; Wu, M.; Zhang, W.; Chen, X.; Zhang, G.; Chen, T.; Liu, G.; et al. Variation in Actinobacterial Community Composition and Potential Function in Different Soil Ecosystems Belonging to the Arid Heihe River Basin of Northwest China. Front. Microbiol. 2019, 10, 2209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shange, R.S.; Ankumah, R.O.; Ibekwe, A.M.; Zabawa, R.; Dowd, S.E. Distinct Soil Bacterial Communities Revealed under a Diversely Managed Agroecosystem. PLoS ONE 2012, 7, e40338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thormann, M.N. The Role of Fungi in Boreal Peatlands. In Boreal Peatland Ecosystems; Wieder, R.K., Vitt, D.H., Eds.; Ecological Studies; Springer: Berlin/Heidelberg, Germany, 2006; pp. 101–123. ISBN 9783540319139. [Google Scholar]
- Artz, R.R.E.; Anderson, I.C.; Chapman, S.J.; Hagn, A.; Schloter, M.; Potts, J.M.; Campbell, C.D. Changes in Fungal Community Composition in Response to Vegetational Succession During the Natural Regeneration of Cutover Peatlands. Microb. Ecol. 2007, 54, 508–522. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, X.; Tian, L.; Ma, L.; Luo, S.; Zhang, J.; Li, X.; Tian, C. Fungal Communities in Ancient Peatlands Developed from Different Periods in the Sanjiang Plain, China. PLoS ONE 2017, 12, e0187575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thormann, M.N. Diversity and Function of Fungi in Peatlands: A Carbon Cycling Perspective. Can. J. Soil Sci. 2006, 86, 281–293. [Google Scholar] [CrossRef]
- Zeilinger, S.; Gupta, V.K.; Dahms, T.E.S.; Silva, R.N.; Singh, H.B.; Upadhyay, R.S.; Gomes, E.V.; Tsui, C.K.-M.; Nayak, S.C. Friends or Foes? Emerging Insights from Fungal Interactions with Plants. FEMS Microbiol. Rev. 2016, 40, 182–207. [Google Scholar] [CrossRef] [PubMed]
- Grube, M.; Wedin, M. Lichenized Fungi and the Evolution of Symbiotic Organization. Microbiol. Spectr. 2016, 4, 4–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, R. Is Groenvlei Really Fed by Groundwater Discharged from the Table Mountain Group (TMG) Aquifer? Water SA 2009, 35. [Google Scholar] [CrossRef] [Green Version]
- Rebelo, A.J.; Emsens, W.-J.; Meire, P.; Esler, K.J. The Impact of Anthropogenically Induced Degradation on the Vegetation and Biochemistry of South African Palmiet Wetlands. Wetl. Ecol. Manag. 2018, 26, 1157–1171. [Google Scholar] [CrossRef]
- Mandiola, S.R.; Grundling, A.T.; Grundling, P.-L.; van der Plicht, J.; van der Waal, B.C.W.; Grootjans, A.P. Ecohydrological Analysis of a South African Through-Flow Mire: Vankervelsvlei Revisited. Mires Peat 2021, 27, 1–14. [Google Scholar] [CrossRef]
- Tian, J.; Huang, X.; Chen, H.; Kang, X.; Wang, Y. Homogeneous Selection Is Stronger for Fungi in Deeper Peat than in Shallow Peat in the Low-Temperature Fens of China. Environ. Res. 2022, 212, 113312. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gao, C.; Liu, H.; Han, D.; Cong, J.; Li, X.; Wang, G. Distribution of Phosphorus Forms in Surface Soils of Typical Peatlands in Northern Great Khingan Mountains and Its Potential to Reconstruct Paleo-Vegetations. J. Environ. Manag. 2022, 302, 114033. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, X.; Duddleston, K.; Hines, M.E. The Effects of PH, Temperature, and Humic-Like Substances on Anaerobic Carbon Degradation and Methanogenesis in Ombrotrophic and Minerotrophic Alaskan Peatlands. Aquat. Geochem. 2020, 26, 221–244. [Google Scholar] [CrossRef]
- Bauer, I.E.; Bhatti, J.S.; Cash, K.J.; Tarnocai, C.; Robinson, S.D. Developing Statistical Models to Estimate the Carbon Density of Organic Soils. Can. J. Soil Sci. 2006, 86, 295–304. [Google Scholar] [CrossRef]
- Lim, A.G.; Loiko, S.V.; Kuzmina, D.M.; Krickov, I.V.; Shirokova, L.S.; Kulizhsky, S.P.; Pokrovsky, O.S. Organic Carbon, and Major and Trace Elements Reside in Labile Low-Molecular Form in the Ground Ice of Permafrost Peatlands: A Case Study of Colloids in Peat Ice of Western Siberia. Environ. Sci. Process. Impacts 2022, 24, 1443–1459. [Google Scholar] [CrossRef]
- Leifeld, J.; Klein, K.; Wüst-Galley, C. Soil Organic Matter Stoichiometry as Indicator for Peatland Degradation. Sci. Rep. 2020, 10, 7634. [Google Scholar] [CrossRef] [PubMed]
- Tipping, E.; Somerville, C.J.; Luster, J. The C:N:P:S Stoichiometry of Soil Organic Matter. Biogeochemistry 2016, 130, 117–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, T.; Ma, Q.; Shi, Q.; Cai, Z.; Zhang, Y.; Cheng, Y.; Nian, H. High Aluminum Stress Drives Different Rhizosphere Soil Enzyme Activities and Bacterial Community Structure between Aluminum-Tolerant and Aluminum-Sensitive Soybean Genotypes. Plant Soil 2019, 440, 409–425. [Google Scholar] [CrossRef]
- Presentato, A.; Piacenza, E.; Turner, R.J.; Zannoni, D.; Cappelletti, M. Processing of Metals and Metalloids by Actinobacteria: Cell Resistance Mechanisms and Synthesis of Metal(Loid)-Based Nanostructures. Microorganisms 2020, 8, 2027. [Google Scholar] [CrossRef] [PubMed]
- Virpiranta, H.; Banasik, M.; Taskila, S.; Leiviskä, T.; Halttu, M.; Sotaniemi, V.-H.; Tanskanen, J. Isolation of Efficient Metal-Binding Bacteria from Boreal Peat Soils and Development of Microbial Biosorbents for Improved Nickel Scavenging. Water 2020, 12, 2000. [Google Scholar] [CrossRef]
- Parvin, S.; Blagodatskaya, E.; Becker, J.N.; Kuzyakov, Y.; Uddin, S.; Dorodnikov, M. Depth Rather than Microrelief Controls Microbial Biomass and Kinetics of C-, N-, P- and S-Cycle Enzymes in Peatland. Geoderma 2018, 324, 67–76. [Google Scholar] [CrossRef]
- Boguta, P.; Skic, K.; Sokołowska, Z.; Frąc, M.; Sas-Paszt, L. Chemical Transformation of Humic Acid Molecules under the Influence of Mineral, Fungal and Bacterial Fertilization in the Context of the Agricultural Use of Degraded Soils. Molecules 2021, 26, 4921. [Google Scholar] [CrossRef] [PubMed]
- Delias, D.S.; Da-Silva, C.J.; Martins, A.C.; de Oliveira, D.S.C.; do Amarante, L. Iron Toxicity Increases Oxidative Stress and Impairs Mineral Accumulation and Leaf Gas Exchange in Soybean Plants during Hypoxia. Environ. Sci. Pollut. Res. 2022, 29, 22427–22438. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Green, S.; Tfaily, M.M.; Prakash, O.; Konstantinidis, K.T.; Corbett, J.E.; Chanton, J.P.; Cooper, W.T.; Kostka, J.E. Microbial Community Structure and Activity Linked to Contrasting Biogeochemical Gradients in Bog and Fen Environments of the Glacial Lake Agassiz Peatland. Appl. Environ. Microbiol. 2012, 78, 7023–7031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Terhonen, E.; Kovalchuk, A.; Tuovila, H.; Chen, H.; Oghenekaro, A.O.; Heinonsalo, J.; Kohler, A.; Kasanen, R.; Vasander, H.; et al. Dominant Tree Species and Soil Type Affect the Fungal Community Structure in a Boreal Peatland Forest. Appl. Environ. Microbiol. 2016, 82, 2632–2643. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxa | Site | Indicator Value | p-Value | Frequency |
|---|---|---|---|---|
| o__Acidimicrobiales.f__C111.g__unidentified | NR | 1 | 0.001 | 6 |
| o__Acidimicrobiales.f__koll13.g__unidentified | NR | 1 | 0.001 | 6 |
| o__Acidimicrobiales.f__AKIW874.g__unidentified | NR | 0.993 | 0.001 | 9 |
| o__Actinomycetales.f__Intrasporangiaceae.Other | NR | 0.991 | 0.001 | 8 |
| o__Acidimicrobiales.f__EB1017.g__unidentified | NR | 0.986 | 0.001 | 8 |
| o__0319.7L14.f__unidentified.g__unidentified | NR | 0.871 | 0.001 | 18 |
| o__Actinomycetales.f__Mycobacteriaceae.g__Mycobacterium | NR | 0.851 | 0.001 | 12 |
| o__Actinomycetales.f__Intrasporangiaceae.g__Janibacter | NR | 0.833 | 0.001 | 5 |
| o__Actinomycetales.f__Frankiaceae.g__Frankia | NR | 0.823 | 0.001 | 6 |
| o__Gaiellales.f__Gaiellaceae.g__unidentified | NR | 0.729 | 0.001 | 18 |
| o__Actinomycetales.f__Micromonosporaceae.g__unidentified | NR | 0.677 | 0.002 | 7 |
| o__Acidimicrobiales.f__TK06.g__unidentified | NR | 0.667 | 0.002 | 4 |
| o__Actinomycetales.f__Pseudonocardiaceae.g__unidentified | NR | 0.667 | 0.001 | 4 |
| o__Nitriliruptorales.f__Nitriliruptoraceae.g__unidentified | NR | 0.667 | 0.003 | 4 |
| o__Rubrobacterales.f__Rubrobacteraceae.g__unidentified | NR | 0.667 | 0.001 | 4 |
| o__Actinomycetales.f__Nocardioidaceae.g__unidentified | NR | 0.570 | 0.019 | 9 |
| o__Actinomycetales.f__Cellulomonadaceae.g__Demequina | NR | 0.5 | 0.007 | 3 |
| o__Actinomycetales.f__Intrasporangiaceae.g__Knoellia | NR | 0.5 | 0.003 | 3 |
| o__Actinomycetales.f__Micromonosporaceae.g__Catellatospora | NR | 0.5 | 0.007 | 3 |
| o__Actinomycetales.f__Micromonosporaceae.g__Pilimelia | NR | 0.5 | 0.008 | 3 |
| o__Micrococcales.f__unidentified.g__unidentified | NR | 0.5 | 0.009 | 3 |
| o__Euzebyales.f__Euzebyaceae.g__Euzebya | NR | 0.5 | 0.009 | 3 |
| o__Actinomycetales.Other.Other | NR | 0.447 | 0.033 | 8 |
| o__Acidimicrobiales.f__Iamiaceae.g__Iamia | NR | 0.333 | 0.038 | 2 |
| o__Actinomycetales.f__Cellulomonadaceae.g__Actinotalea | NR | 0.333 | 0.043 | 2 |
| o__Actinomycetales.f__Cryptosporangiaceae.g__unidentified | NR | 0.333 | 0.043 | 2 |
| o__Actinomycetales.f__Frankiaceae.Other | NR | 0.333 | 0.048 | 2 |
| o__Actinomycetales.f__Micromonosporaceae.g__Dactylosporangium | NR | 0.333 | 0.041 | 2 |
| o__Actinomycetales.f__Pseudonocardiaceae.Other | NR | 0.333 | 0.039 | 2 |
| o__Solirubrobacterales.f__Conexibacteraceae.g__Conexibacter | GK | 0.993 | 0.001 | 13 |
| o__Acidimicrobiales.f__unidentified.g__unidentified | GK | 0.965 | 0.001 | 21 |
| o__Actinomycetales.f__unidentified.g_unidentified | GK | 0.913 | 0.001 | 19 |
| o__Solirubrobacterales.f__Conexibacteraceae.g_unidentified | GK | 0.849 | 0.001 | 12 |
| o__Actinomycetales.f__Actinosynnemataceae.Other | GK | 0.775 | 0.001 | 8 |
| o__Solirubrobacterales.Other.Other | GK | 0.667 | 0.003 | 6 |
| o__Actinomycetales.f__Actinospicaceae.g_unidentified | GK | 0.664 | 0.001 | 7 |
| o__Solirubrobacterales.f__unidentified.g_unidentified | GK | 0.598 | 0.015 | 19 |
| o__Solirubrobacterales.f__Conexibacteraceae.Other | GK | 0.556 | 0.005 | 5 |
| o__Actinomycetales.f__Frankiaceae.g_unidentified | GK | 0.505 | 0.01 | 7 |
| o__Actinomycetales.f__Micrococcaceae.g__Sinomonas | GK | 0.444 | 0.025 | 4 |
| o__Actinomycetales.f__Nocardiaceae.g__Nocardia | GK | 0.444 | 0.021 | 4 |
| o__Actinomycetales.f__Actinosynnemataceae.g_unidentified | GK | 0.333 | 0.027 | 3 |
| o__Actinomycetales.f__Microbacteriaceae.g__Frigoribacterium | VV | 0.583 | 0.006 | 7 |
| o__Actinomycetales.f__Corynebacteriaceae.g__Corynebacterium | VV | 0.5 | 0.017 | 6 |
| o__Actinomycetales.f__Propionibacteriaceae.g__Propionibacterium | VV | 0.496 | 0.026 | 7 |
| Taxa | Site | Indicator Value | p-Value | Frequency |
| o__Eurotiales.f__Trichocomaceae.g__Penicillium | NR | 0.846 | 0.014 | 20 |
| o__Pleosporales.f__Venturiaceae.g__unidentified | NR | 0.666 | 0.006 | 8 |
| o__Pleosporales.f__Incertae_sedis.g__Phoma | GK | 1 | 0.001 | 12 |
| o__Eurotiales.f__Trichocomaceae.g__unidentified | GK | 0.881 | 0.002 | 9 |
| o__Eurotiales.f__Trichocomaceae.g__Talaromyces | GK | 0.858 | 0.001 | 9 |
| o__Archaeorhizomycetales.f__Archaeorhizomycetaceae.g__Archaeorhizomyces | GK | 0.833 | 0.001 | 5 |
| o__Pleosporales.f__Venturiaceae.g__Anungitopsis | GK | 0.667 | 0.004 | 4 |
| o__Pleosporales.f__unidentified.g__unidentified | GK | 0.651 | 0.013 | 12 |
| o__Capnodiales.f__Incertae_sedis.g__Capnobotryella | GK | 0.333 | 0.05 | 2 |
| o__Capnodiales.f__Mycosphaerellaceae.g__Septoria | VV | 0.889 | 0.001 | 8 |
| o__Pleosporales.f__Phaeosphaeriaceae.g__Ampelomyces | VV | 0.646 | 0.017 | 8 |
| o__Dothideales.f__Dothioraceae.g__Aureobasidium | VV | 0.619 | 0.017 | 8 |
| Nuwejaars River System (NR) Developing Peatland | Goukou River System (GK) Impacted Peatland | |||
|---|---|---|---|---|
| Actinobacteria | Fungi | Actinobacteria | Fungi | |
| Goukou River system (GK) | 0.995 *** | 0.762 *** | - | - |
| Vankervelsvlei (VV; undisturbed peatland) | 0.436 *** | 0.997 *** | 0.650 *** | 0.912 *** |
| 580–600 cm | 780–800 cm | 980–1000 cm | |
|---|---|---|---|
| 780–800 cm | 0.111 | ||
| 980–1000 cm | 0.074 | 0.185 | |
| 1180–2000 cm | 0.889 * | 0.704 * | 0.889 * |
| Trophic Mode | Ascomycete Taxon | Basidiomycete Taxon |
|---|---|---|
| Pathotroph–Saprotroph–Symbiotroph | Acremonium, Alternaria, Aureobasidium, Candida, Epicoccum, Fusarium, Phoma, Trichoderma | Agaricales, Cryptococcus |
| Pathotroph–Saprotroph | Pleospora, Teratosphaeriaceae | Rhodotorula |
| Pathotroph–Symbiotroph | Tricholomataceae | - |
| Saprotroph–Symbiotroph | Ampelomyces, Myxotrichaceae | - |
| Saprotroph | Eupenicillium, Lulwoana, Penicillium | Termitomyces |
| Pathotroph | Haematonectria, Lecanicillium, Lewia, Metarhizium | - |
| Symbiotroph | Lecythophora, Tuberaceae | Cortinarius |
| Sampling Site | Abundant Fungal Taxa | Fungal Phylum, Order | Fungal Guild |
|---|---|---|---|
| GK | Beauveria | Ascomycota, Sordariomycetes | Animal pathogen |
| Chroogomphus | Basidiomycota, Agaricomycetes | Ectomycorrhizal, fungal parasite | |
| Cortinarius | Basidiomycota, Agaricomycetes | Ectomycorrhizal | |
| Diaportharles | Ascomycota, Sordariomycetes | Endophyte, plant pathogen, undefined saprotroph | |
| Penicillium | Ascomycota, Eurotiomycetes | Dung saprotroph, undefined saprotroph, wood saprotroph | |
| Trichocomaceae | Ascomycota, Eurotiomycetes | Saprotroph | |
| Tricholomataceae | Basidiomycota Agaricomycetes | Ectomycorrhizal, fungal parasite | |
| Venturiaceae | Ascomycota, Dothideomycetes | Endophyte, plant pathogen, undefined saprotroph | |
| NR | Acremonium | Ascomycota, Sordariomycetes | Animal pathogen, endophyte, fungal parasite, plant pathogen, wood saprotroph |
| Epicoccum | Ascomycota, Dothideomycetes | Endophyte, fungal parasite, lichen parasite, plant pathogen, wood saprotroph | |
| Geoglossum | Ascomycota, Leotiomycetes | Undefined saprotroph | |
| Haematonectria | Ascomycota, Sodariomycetes | Plant pathogen | |
| Lulwoana | Ascomycota, Sordariomycetes | Undefined saprotroph | |
| Phoma | Ascomycota, Dothideomycetes | Endophyte, dung saprotroph, lichen parasite, litter saprotroph, plant pathogen, soil saprotroph, wood saprotroph | |
| Trichocomaceae | Ascomycota, Eurotiomycetes | Saprotroph | |
| VV | Candida | Ascomycota, Saccharomycetes | Animal pathogen, endophyte, epiphyte, endosymbiont, soil saprotroph, undefined saprotroph |
| Cryptococcus | Basidiomycota, Tremellomycetes | Animal pathogen, endophyte, epiphyte, undefined saprotroph | |
| Epicoccum | Ascomycota, Dothideomycetes | Endophyte, fungal parasite, lichen parasite, plant pathogen, wood saprotroph | |
| Leucosporidiales | Basidiomycota, Microbotryomycetes | Soil saprotroph, undefined saprotroph | |
| Rhodosporidium | Basidiomycota, Microbotryomycetes | Undefined saprotroph | |
| Tremella | Basidiomycota, Tremellomycetes | Saprotroph | |
| Trichoderma | Ascomycota, Sordariomycetes | Animal pathogen, endophyte, epiphyte, fungal parasite, plant pathogen, wood saprotroph |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weels, S.S.L.; Welz, P.J.; Prins, A.; Le Roes-Hill, M. Impact of Physicochemical Parameters on the Diversity and Distribution of Microbial Communities Associated with Three South African Peatlands. Microorganisms 2022, 10, 2103. https://doi.org/10.3390/microorganisms10112103
Weels SSL, Welz PJ, Prins A, Le Roes-Hill M. Impact of Physicochemical Parameters on the Diversity and Distribution of Microbial Communities Associated with Three South African Peatlands. Microorganisms. 2022; 10(11):2103. https://doi.org/10.3390/microorganisms10112103
Chicago/Turabian StyleWeels, Shandré S. L., Pamela J. Welz, Alaric Prins, and Marilize Le Roes-Hill. 2022. "Impact of Physicochemical Parameters on the Diversity and Distribution of Microbial Communities Associated with Three South African Peatlands" Microorganisms 10, no. 11: 2103. https://doi.org/10.3390/microorganisms10112103