
Late-Stage Functionalisation of Polycyclic (N-Hetero-) Aromatic Hydrocarbons by Detoxifying CYP5035S7 Monooxygenase of the White-Rot Fungus Polyporus arcularius


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
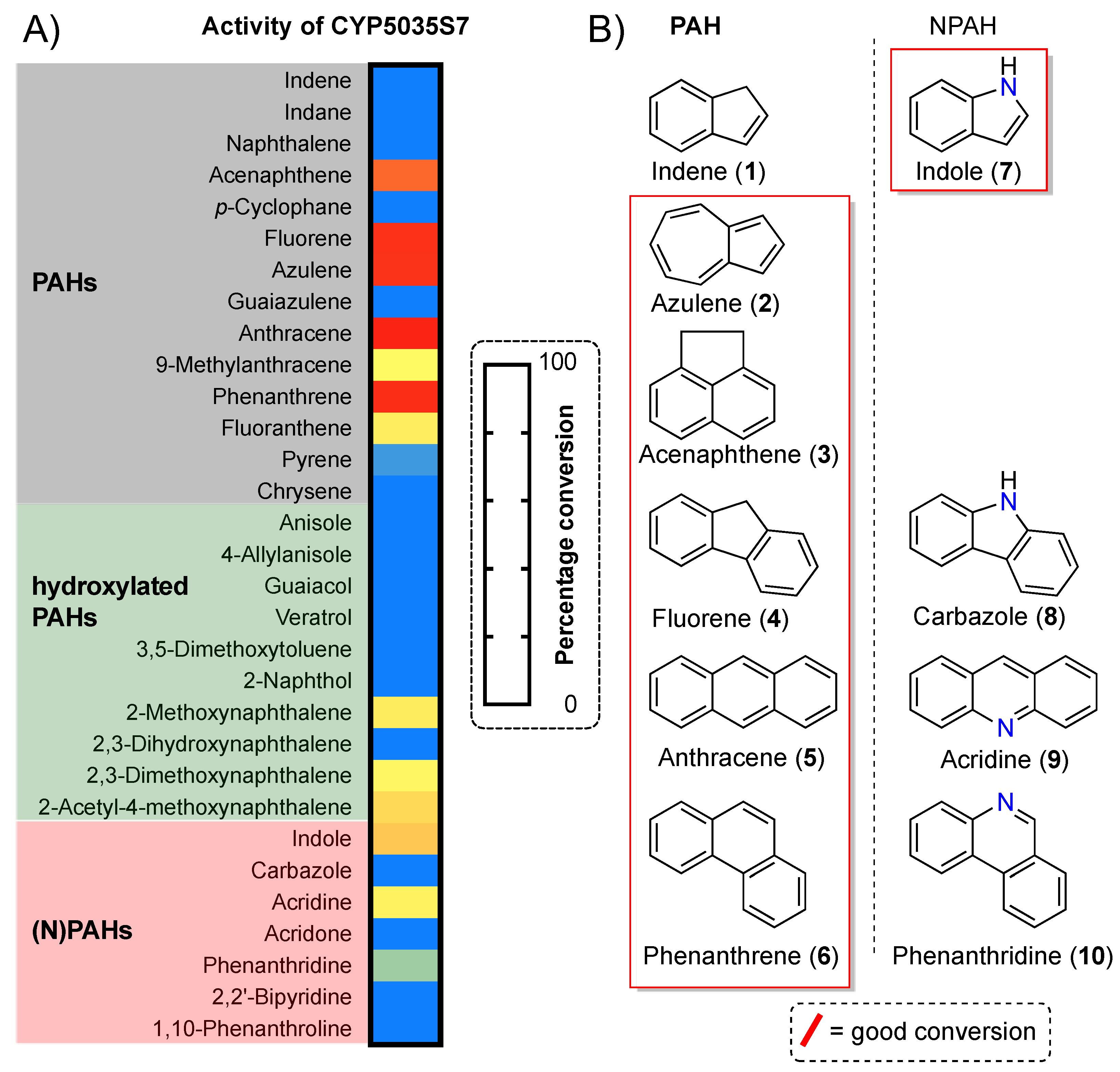
2.1. Substrate Screening
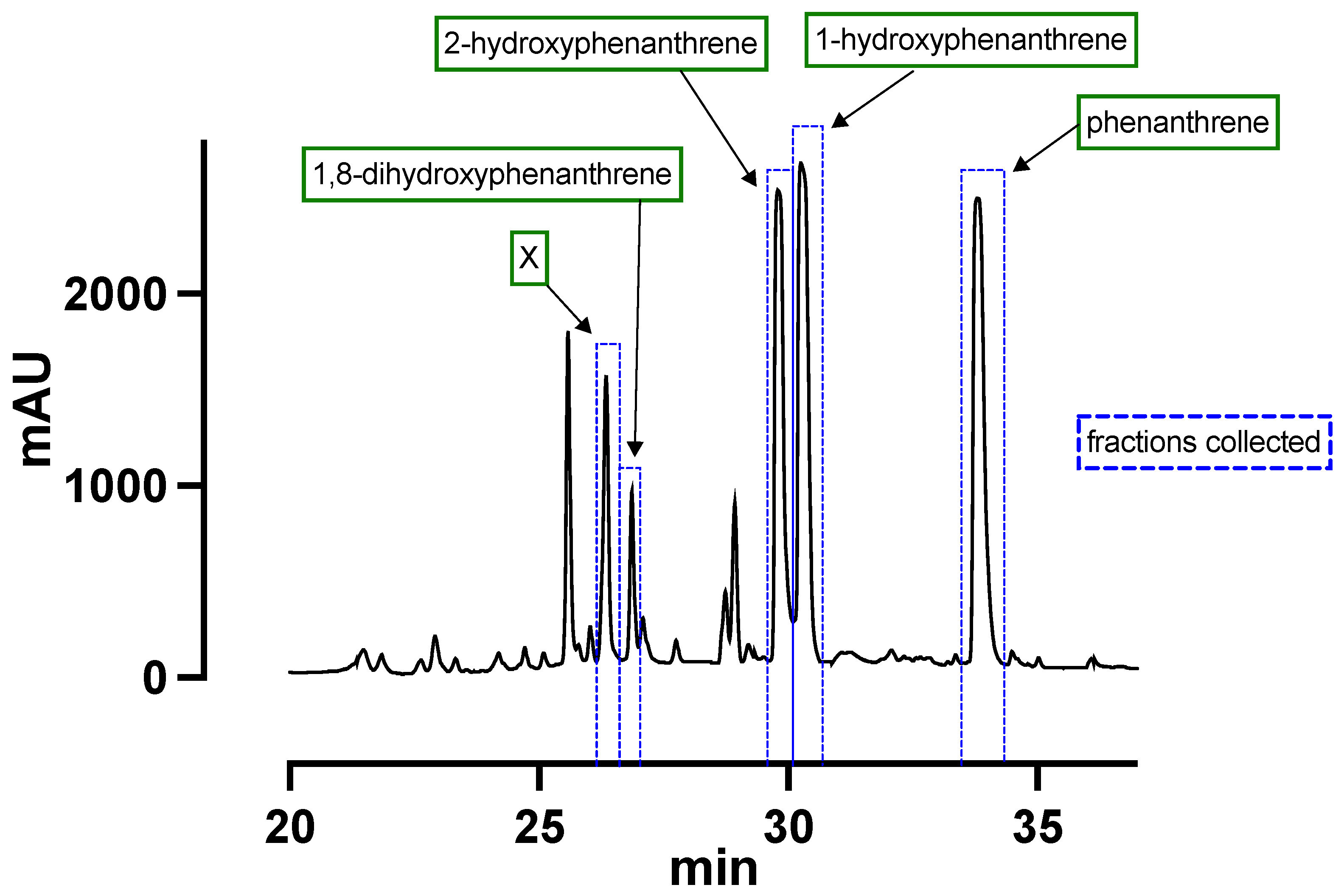
2.2. Product Isolation
2.3. NMR of Products
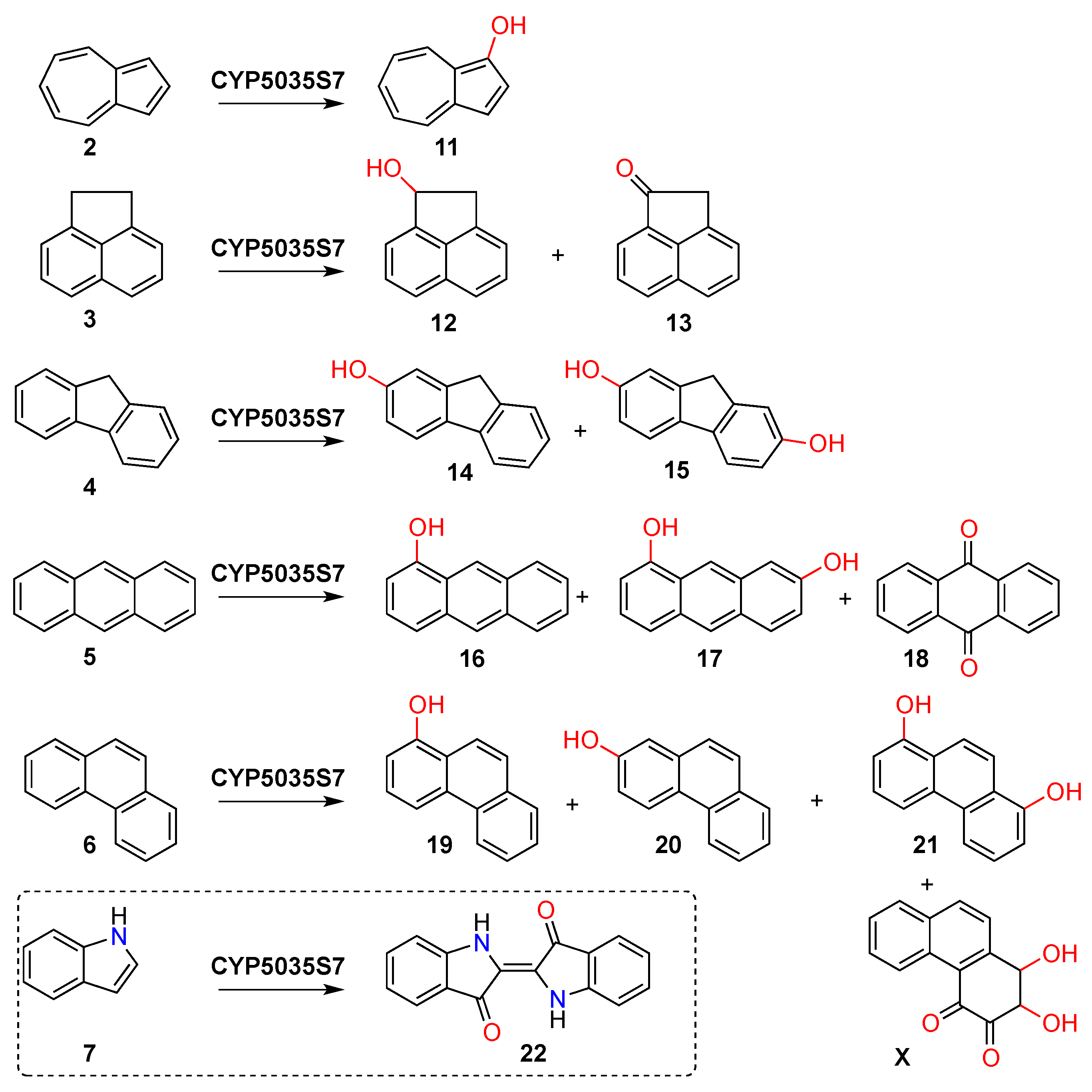
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Idowu, O.; Semple, K.T.; Ramadass, K.; O’Connor, W.; Hansbro, P.; Thavamani, P. Beyond the obvious: Environmental health implications of polar polycyclic aromatic hydrocarbons. Environ. Int. 2019, 123, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K. Chemistry of Polycyclic Aromatic Hydrocarbons (PAHs), Nitropolycyclic Aromatic Hydrocarbons (NPAHs) and Other Oxidative Derivatives of PAHs. In Polycyclic Aromatic Hydrocarbons; Springer: Singapore, 2018; pp. 3–10. ISBN 9789811067754. [Google Scholar]
- Aumaitre, C.; Morin, J. Polycyclic Aromatic Hydrocarbons as Potential Building Blocks for Organic Solar Cells. Chem. Rec. 2019, 19, 1142–1154. [Google Scholar] [CrossRef]
- Okamoto, H. Organic chemistry of π-conjugated polycyclic aromatic hydrocarbons: Acenes and phenacenes. In Physics and Chemistry of Carbon-Based Materials: Basics and Applications; Kubozono, Y., Ed.; Springer Nature Singapore Pte Ltd.: Singapore, 2019; pp. 211–228. ISBN 9789811334177. [Google Scholar]
- Davidson-Hall, T.; Kajiyama, Y.; Aziz, H. Organic Light Emitting Device Materials for Displays. In Materials for Solid State Lighting and Displays; Kitai, A., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2016; Volume 2, pp. 183–230. [Google Scholar]
- Pisula, W.; Feng, X.; Müllen, K. Tuning the Columnar Organization of Discotic Polycyclic Aromatic Hydrocarbons. Adv. Mater. 2010, 22, 3634–3649. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Wang, H. HOMO-LUMO energy splitting in polycyclic aromatic hydrocarbons and their derivatives. Proc. Combust. Inst. 2019, 37, 953–959. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Ehrat, F.; Urban, P.; Teves, R.; Wyrwich, R.; Döblinger, M.; Feldmann, J.; Urban, A.S.; Stolarczyk, J.K. Effect of nitrogen atom positioning on the trade-off between emissive and photocatalytic properties of carbon dots. Nat. Commun. 2017, 8, 1401. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Monteiro, J.H.S.K.; de Bettencourt-Dias, A.; Catalano, V.J.; Chalifoux, W.A. Pyrenes, Peropyrenes, and Teropyrenes: Synthesis, Structures, and Photophysical Properties. Angew. Chem. 2016, 128, 10583–10586. [Google Scholar] [CrossRef]
- Liu, J.; Feng, X. Bottom-Up Synthesis of Nitrogen-Doped Polycyclic Aromatic Hydrocarbons. Synlett 2020, 31, 211–222. [Google Scholar] [CrossRef]
- Hagui, W.; Doucet, H.; Soulé, J.F. Application of Palladium-Catalyzed C(sp2)–H Bond Arylation to the Synthesis of Polycyclic (Hetero)Aromatics. Chem 2019, 5, 2006–2078. [Google Scholar] [CrossRef]
- Wencel-Delord, J.; Glorius, F. C-H bond activation enables the rapid construction and late-stage diversification of functional molecules. Nat. Chem. 2013, 5, 369–375. [Google Scholar] [CrossRef]
- Kancherla, S.; Jørgensen, K.; Fernández-Ibáñez, M. Recent Developments in Palladium-Catalysed Non-Directed C–H Bond Activation in Arenes. Synthesis 2019, 51, 643–663. [Google Scholar] [CrossRef]
- Mathew, B.P.; Kuram, M.R. Emerging C H functionalization strategies for constructing fused polycyclic aromatic hydrocarbons and nanographenes. Inorg. Chim. Acta 2019, 490, 112–129. [Google Scholar] [CrossRef]
- Fessner, N.D. P450 Monooxygenases Enable Rapid Late-Stage Diversification of Natural Products via C−H Bond Activation. ChemCatChem 2019, 11, 2226–2242. [Google Scholar] [CrossRef] [Green Version]
- Takikawa, H.; Nishii, A.; Sakai, T.; Suzuki, K. Aryne-based strategy in the total synthesis of naturally occurring polycyclic compounds. Chem. Soc. Rev. 2018, 47, 8030–8056. [Google Scholar] [CrossRef]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef] [PubMed]
- Cernak, T.; Dykstra, K.D.; Tyagarajan, S.; Vachal, P.; Krska, S.W. The medicinal chemist’s toolbox for late stage functionalization of drug-like molecules. Chem. Soc. Rev. 2016, 45, 546–576. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, J.B.; Selby, A.L.; Freeman, J.P.; Evans, F.E.; Cerniglia, C.E. Metabolism of phenanthrene by Phanerochaete chrysosporium. Appl. Environ. Microbiol. 1991, 57, 3310–3316. [Google Scholar] [CrossRef] [Green Version]
- Syed, K.; Doddapaneni, H.; Subramanian, V.; Lam, Y.W.; Yadav, J.S. Genome-to-function characterization of novel fungal P450 monooxygenases oxidizing polycyclic aromatic hydrocarbons (PAHs). Biochem. Biophys. Res. Commun. 2010, 399, 492–497. [Google Scholar] [CrossRef] [Green Version]
- Hirosue, S.; Tazaki, M.; Hiratsuka, N.; Yanai, S.; Kabumoto, H.; Shinkyo, R.; Arisawa, A.; Sakaki, T.; Tsunekawa, H.; Johdo, O.; et al. Insight into functional diversity of cytochrome P450 in the white-rot basidiomycete Phanerochaete chrysosporium: Involvement of versatile monooxygenase. Biochem. Biophys. Res. Commun. 2011, 407, 118–123. [Google Scholar] [CrossRef]
- Syed, K.; Shale, K.; Pagadala, N.S.; Tuszynski, J. Systematic Identification and Evolutionary Analysis of Catalytically Versatile Cytochrome P450 Monooxygenase Families Enriched in Model Basidiomycete Fungi. PLoS ONE 2014, 9, e86683. [Google Scholar] [CrossRef]
- Sakai, K.; Matsuzaki, F.; Wise, L.; Sakai, Y.; Jindou, S.; Ichinose, H.; Takaya, N.; Kato, M.; Wariishi, H.; Shimizu, M. Biochemical Characterization of CYP505D6, a Self-Sufficient Cytochrome P450 from the White-Rot Fungus Phanerochaete chrysosporium. Appl. Environ. Microbiol. 2018, 84, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Syed, K.; Porollo, A.; Lam, Y.W.; Grimmett, P.E.; Yadav, J.S. CYP63A2, a Catalytically Versatile Fungal P450 Monooxygenase Capable of Oxidizing Higher-Molecular-Weight Polycyclic Aromatic Hydrocarbons, Alkylphenols, and Alkanes. Appl. Environ. Microbiol. 2013, 79, 2692–2702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syed, K.; Porollo, A.; Miller, D.; Yadav, J.S. Rational engineering of the fungal P450 monooxygenase CYP5136A3 to improve its oxidizing activity toward polycyclic aromatic hydrocarbons. Protein Eng. Des. Sel. 2013, 26, 553–557. [Google Scholar] [CrossRef]
- Syed, K.; Porollo, A.; Lam, Y.W.; Yadav, J.S. A Fungal P450 (CYP5136A3) Capable of Oxidizing Polycyclic Aromatic Hydrocarbons and Endocrine Disrupting Alkylphenols: Role of Trp129 and Leu324. PLoS ONE 2011, 6, e28286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chigu, N.L.; Hirosue, S.; Nakamura, C.; Teramoto, H.; Ichinose, H.; Wariishi, H. Cytochrome P450 monooxygenases involved in anthracene metabolism by the white-rot basidiomycete Phanerochaete chrysosporium. Appl. Microbiol. Biotechnol. 2010, 87, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Fessner, N.D.; Nelson, D.R.; Glieder, A. Evolution and enrichment of CYP5035 in Polyporales: Functionality of an understudied P450 family. Appl. Microbiol. Biotechnol. 2021, 105, 6779–6792. [Google Scholar] [CrossRef]
- Weis, R.; Luiten, R.; Skranc, W.; Schwab, H.; Wubbolts, M.; Glieder, A. Reliable high-throughput screening with by limiting yeast cell death phenomena. FEMS Yeast Res. 2004, 5, 179–189. [Google Scholar] [CrossRef] [Green Version]
- Asao, T.; Ito, S.; Morita, N. 1-Hydroxyazulene and 3-hydroxyguaiazulene: Synthesis and their properties. Tetrahedron Lett. 1989, 30, 6693–6696. [Google Scholar] [CrossRef]
- Pothuluri, J.V.; Freeman, J.P.; Evans, F.E.; Cerniglia, C.E. Fungal metabolism of acenaphthene by Cunninghamella elegans. Appl. Environ. Microbiol. 1992, 58, 3654–3659. [Google Scholar] [CrossRef] [Green Version]
- Abraham, R.J.; Canton, M.; Reid, M.; Griffiths, L. Proton chemical shifts in NMR. Part 14. Proton chemical shifts, ring currents and π electron effects in condensed aromatic hydrocarbons and substituted benzenes. J. Chem. Soc. Perkin Trans. 2000, 2, 803–812. [Google Scholar] [CrossRef]
- Salvador, M.A.; Coelho, P.J.; Burrows, H.D.; Oliveira, M.M.; Carvalho, L.M. Studies under Continuous Irradiation of Photochromic Spiro[fluorenopyran-thioxanthenes]. Helv. Chim. Acta 2004, 87, 1400–1410. [Google Scholar] [CrossRef] [Green Version]
- Finkelstein, Z.I.; Baskunov, B.P.; Golovlev, E.L.; Vervoort, J.; Rietjens, I.M.C.M.; Baboshin, M.A.; Golovleva, L.A. Fluorene Transformation by Bacteria of the Genus Rhodococcus. Microbiology 2003, 72, 660–665. [Google Scholar] [CrossRef]
- Kageyama, T.; Koizumi, Y.; Igarashi, T.; Sakurai, T. 1-(Arylmethyloxy)anthracenes: How substituents affect their photoreactivity and ability to initiate radical and cationic polymerizations. Polym. J. 2012, 44, 1022–1029. [Google Scholar] [CrossRef] [Green Version]
- Coleman, R.S.; Mortensen, M.A. Stereocontrolled synthesis of anthracene β-C-ribosides: Fluorescent probes for photophysical studies of DNA. Tetrahedron Lett. 2003, 44, 1215–1219. [Google Scholar] [CrossRef]
- 1H-NMR-Spektroskopie. Laborpraxis Band 4: Analytische Methoden; Aprentas, Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 181–242. ISBN 978-3-0348-0971-9. [Google Scholar]
- Wu, A.; Duan, Y.; Xu, D.; Penning, T.M.; Harvey, R.G. Regiospecific oxidation of polycyclic aromatic phenols to quinones by hypervalent iodine reagents. Tetrahedron 2010, 66, 2111–2118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Silva, M.; Esposito, E.; Moody, J.D.; Canhos, V.P.; Cerniglia, C.E. Metabolism of aromatic hydrocarbons by the filamentous fungus Cyclothyrium sp. Chemosphere 2004, 57, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Sack, U.; Heinze, T.M.; Deck, J.; Cerniglia, C.E.; Martens, R.; Zadrazil, F.; Fritsche, W. Comparison of phenanthrene and pyrene degradation by different wood-decaying fungi. Appl. Environ. Microbiol. 1997, 63, 3919–3925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fessner, N.D.; Srdič, M.; Weber, H.; Schmid, C.; Schönauer, D.; Schwaneberg, U.; Glieder, A. Preparative-Scale Production of Testosterone Metabolites by Human Liver Cytochrome P450 Enzyme 3A4. Adv. Synth. Catal. 2020, 362, 2725–2738. [Google Scholar] [CrossRef]
- Syed, K.; Yadav, J.S. P450 monooxygenases (P450ome) of the model white rot fungus Phanerochaete chrysosporium. Crit. Rev. Microbiol. 2012, 38, 339–363. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Couto, S. Industrial and environmental applications of white-rot fungi. Mycosphere 2017, 8, 456–466. [Google Scholar] [CrossRef]
- Ellouze, M.; Sayadi, S. White-Rot Fungi and their Enzymes as a Biotechnological Tool for Xenobiotic Bioremediation. In Management of Hazardous Wastes; InTech: London, UK, 2016. [Google Scholar]
- Tortella, G.; Durán, N.; Rubilar, O.; Parada, M.; Diez, M.C. Are white-rot fungi a real biotechnological option for the improvement of environmental health? Crit. Rev. Biotechnol. 2015, 35, 165–172. [Google Scholar] [CrossRef]
- Chen, B.; Wang, Y.; Hu, D. Biosorption and biodegradation of polycyclic aromatic hydrocarbons in aqueous solutions by a consortium of white-rot fungi. J. Hazard. Mater. 2010, 179, 845–851. [Google Scholar] [CrossRef]
- Gao, D.; Du, L.; Yang, J.; Wu, W.-M.; Liang, H. A critical review of the application of white rot fungus to environmental pollution control. Crit. Rev. Biotechnol. 2010, 30, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Dannert, C. Biocatalytic portfolio of Basidiomycota. Curr. Opin. Chem. Biol. 2016, 31, 40–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandini, M. Electrophilicity: The “dark-side” of indole chemistry. Org. Biomol. Chem. 2013, 11, 5206. [Google Scholar] [CrossRef] [PubMed]
- Ottoni, O.; Neder, A.; Dias, A.K.B.; Cruz, R.P.A.; Aquino, L.B. Acylation of Indole under Friedel−Crafts ConditionsAn Improved Method To Obtain 3-Acylindoles Regioselectively. Org. Lett. 2001, 3, 1005–1007. [Google Scholar] [CrossRef]
- Szatylowicz, H.; Jezuita, A.; Krygowski, T.M. On the relations between aromaticity and substituent effect. Struct. Chem. 2019, 30, 1529–1548. [Google Scholar] [CrossRef] [Green Version]
- Estévez-Fregoso, M.; Hernández-Trujillo, J. Electron delocalization and electron density of small polycyclic aromatic hydrocarbons in singlet excited states. Phys. Chem. Chem. Phys. 2016, 18, 11792–11799. [Google Scholar] [CrossRef] [PubMed]
- Solà, M. Forty years of Clar’s aromatic π-sextet rule. Front. Chem. 2013, 1, 4–11. [Google Scholar] [CrossRef] [Green Version]
- Titaley, I.A.; Walden, D.M.; Dorn, S.E.; Ogba, O.M.; Massey Simonich, S.L.; Cheong, P.H.-Y. Evaluating Computational and Structural Approaches to Predict Transformation Products of Polycyclic Aromatic Hydrocarbons. Environ. Sci. Technol. 2019, 53, 1595–1607. [Google Scholar] [CrossRef]
- Pabulo, H. Rampelotto Grand Challenges in Fungal Biotechnology; Nevalainen, H., Ed.; Grand Challenges in Biology and Biotechnology; Springer International Publishing: Cham, Switzerland, 2020; ISBN 978-3-030-29540-0. [Google Scholar]
- Xin, H.; Gao, X. Application of Azulene in Constructing Organic Optoelectronic Materials: New Tricks for an Old Dog. Chempluschem 2017, 82, 945–956. [Google Scholar] [CrossRef] [Green Version]
- de Carvalho, C.C.C.R. Whole cell biocatalysts: Essential workers from Nature to the industry. Microb. Biotechnol. 2017, 10, 250–263. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Fernández-Fueyo, E.; Hollmann, F.; Paul, C.E.; Pesic, M.; Schmidt, S.; Wang, Y.; Younes, S.; Zhang, W. Biocatalytic Oxidation Reactions: A Chemist’s Perspective. Angew. Chemie Int. Ed. 2018, 57, 9238–9261. [Google Scholar] [CrossRef] [PubMed]
- Kratzer, R.; Woodley, J.M.; Nidetzky, B. Rules for biocatalyst and reaction engineering to implement effective, NAD(P)H-dependent, whole cell bioreductions. Biotechnol. Adv. 2015, 33, 1641–1652. [Google Scholar] [CrossRef] [Green Version]
- Vogl, T.; Glieder, A. Regulation of Pichia pastoris promoters and its consequences for protein production. New Biotechnol. 2013, 30, 385–404. [Google Scholar] [CrossRef] [PubMed]
- Weninger, A.; Glieder, A.; Vogl, T. A toolbox of endogenous and heterologous nuclear localization sequences for the methylotrophic yeast Pichia pastoris. FEMS Yeast Res. 2015, 15, fov082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, A.N.; Singh, S.; Mishra, S.; Gupta, A. Recent Advancement in White Biotechnology Through Fungi; Fungal Biology; Springer International Publishing: Cham, Switzerland, 2019; ISBN 978-3-030-25505-3. [Google Scholar]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fessner, N.D.; Grimm, C.; Kroutil, W.; Glieder, A. Late-Stage Functionalisation of Polycyclic (N-Hetero-) Aromatic Hydrocarbons by Detoxifying CYP5035S7 Monooxygenase of the White-Rot Fungus Polyporus arcularius. Biomolecules 2021, 11, 1708. https://doi.org/10.3390/biom11111708
Fessner ND, Grimm C, Kroutil W, Glieder A. Late-Stage Functionalisation of Polycyclic (N-Hetero-) Aromatic Hydrocarbons by Detoxifying CYP5035S7 Monooxygenase of the White-Rot Fungus Polyporus arcularius. Biomolecules. 2021; 11(11):1708. https://doi.org/10.3390/biom11111708
Chicago/Turabian StyleFessner, Nico D., Christopher Grimm, Wolfgang Kroutil, and Anton Glieder. 2021. "Late-Stage Functionalisation of Polycyclic (N-Hetero-) Aromatic Hydrocarbons by Detoxifying CYP5035S7 Monooxygenase of the White-Rot Fungus Polyporus arcularius" Biomolecules 11, no. 11: 1708. https://doi.org/10.3390/biom11111708