

Development of Simple Sequence Repeat Markers and Genetic Diversity Evaluation of Mycocentrospora acerina in Yunnan Province, China
Abstract
:1. Introduction
2. Materials and Methods
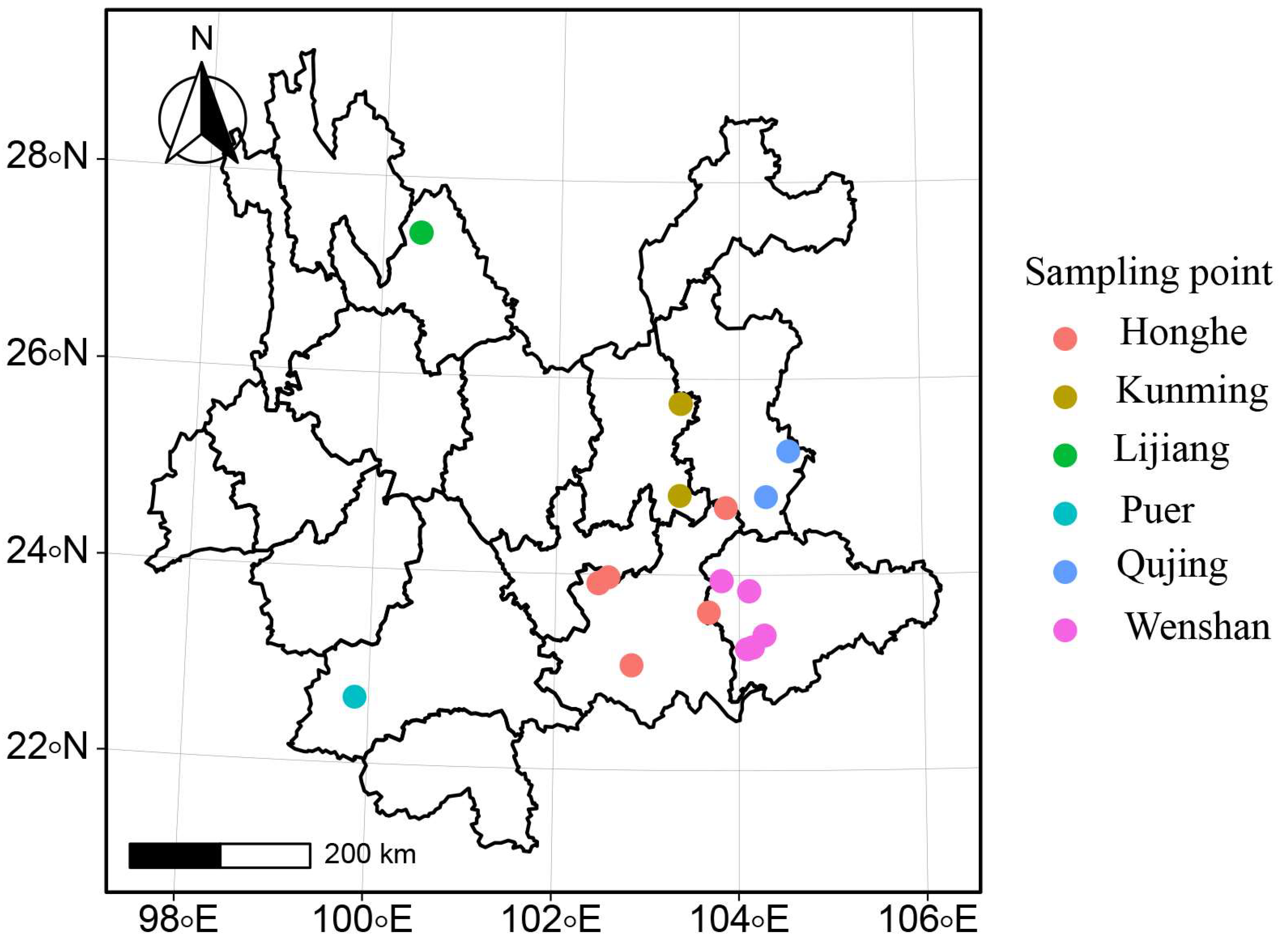
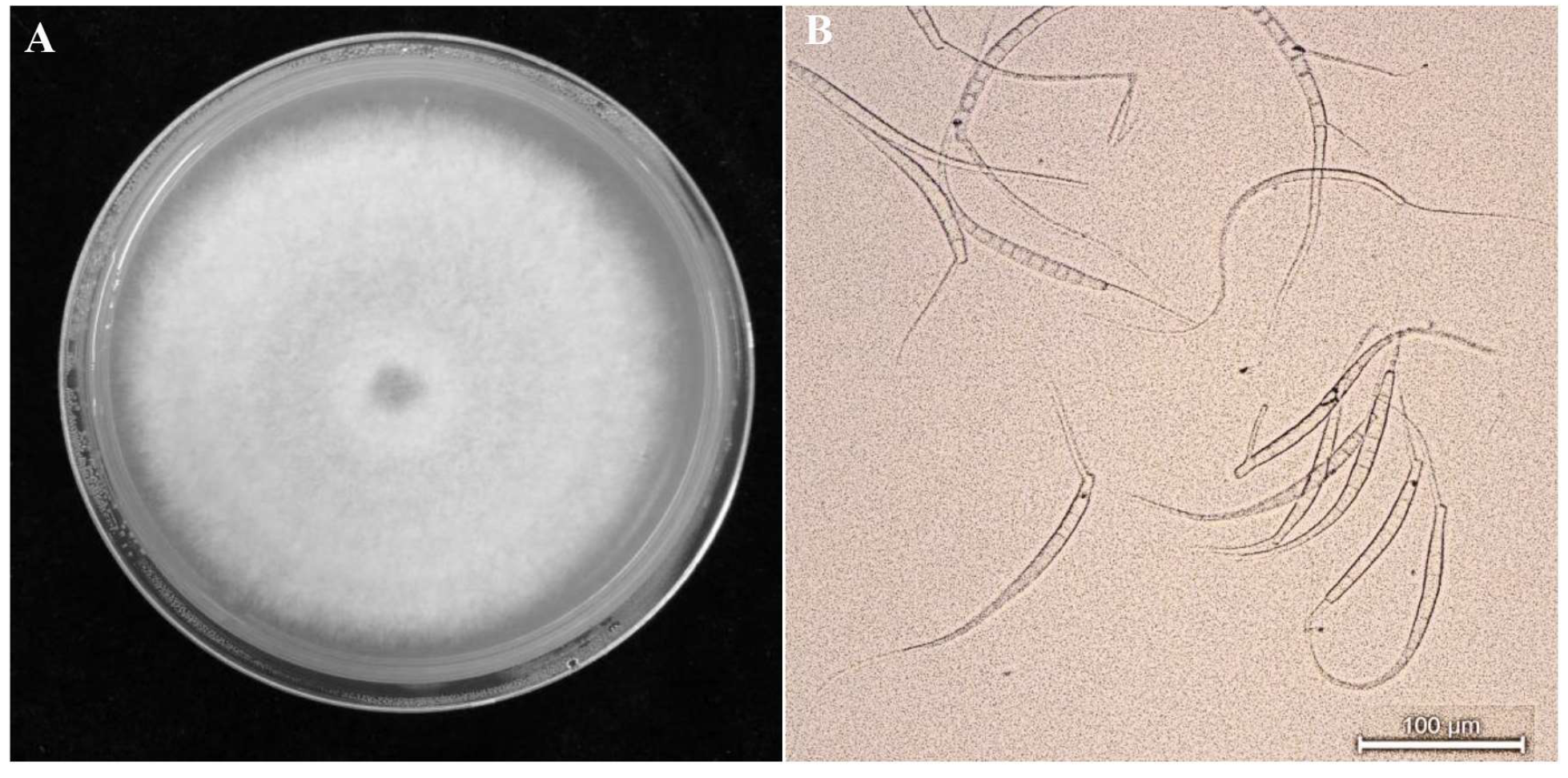
2.1. Strain Isolation and Observation
2.2. DNA Extraction and Polymerase Chain Reaction Amplification of Internal Transcribed Spacer (ITS1/4) Regions
2.3. Simple Sequence Repeat Screening
2.4. Primer Design
2.5. SSR Analysis of Mycocentrospora acerina Genome
2.6. Statistical Analysis
3. Results
3.1. Isolation and Identification of Strains
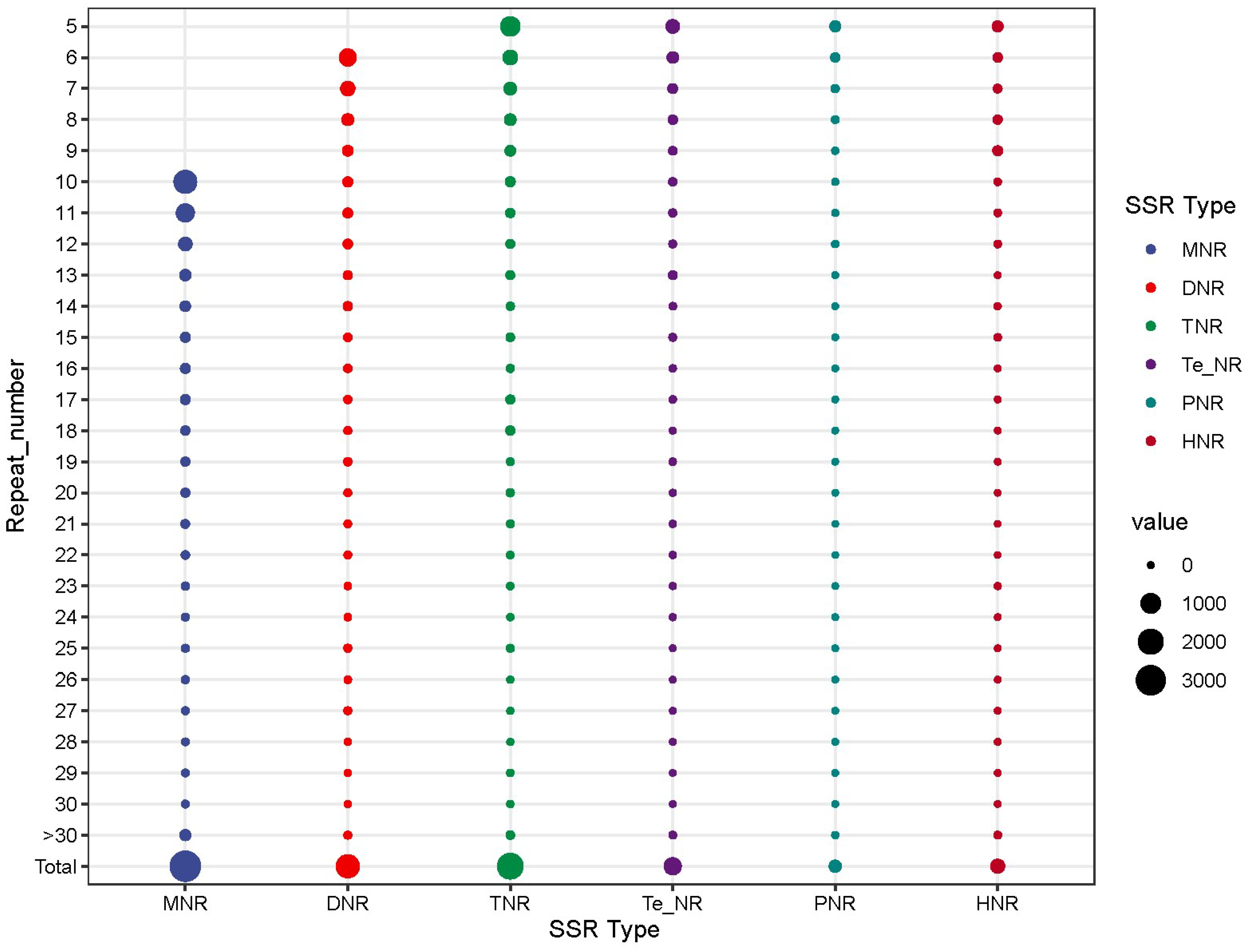
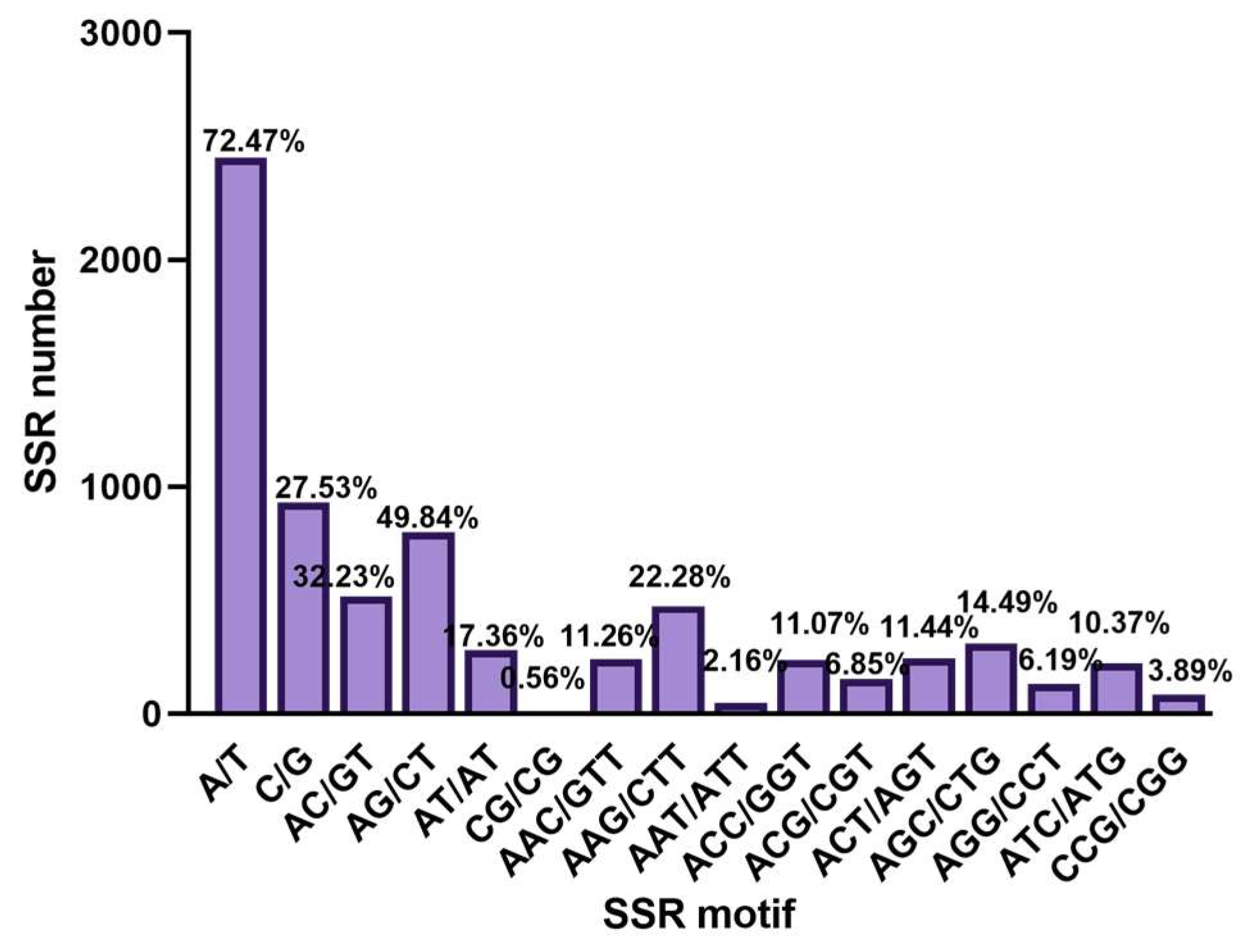
3.2. Genomic SSR Analysis
3.3. Polymorphism of SSR Primers
3.4. Genetic Diversity within Populations
3.5. Diversity between Populations
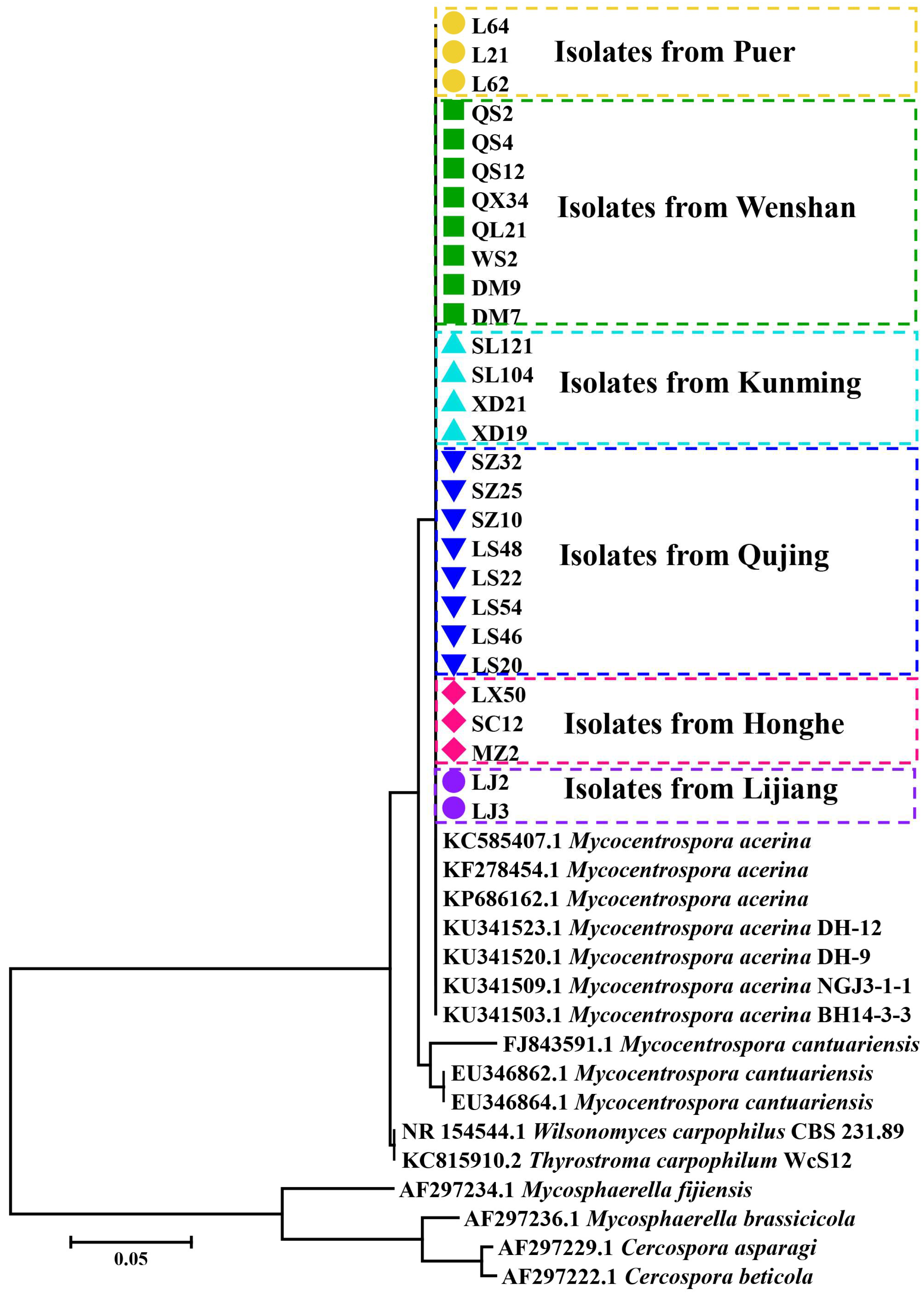
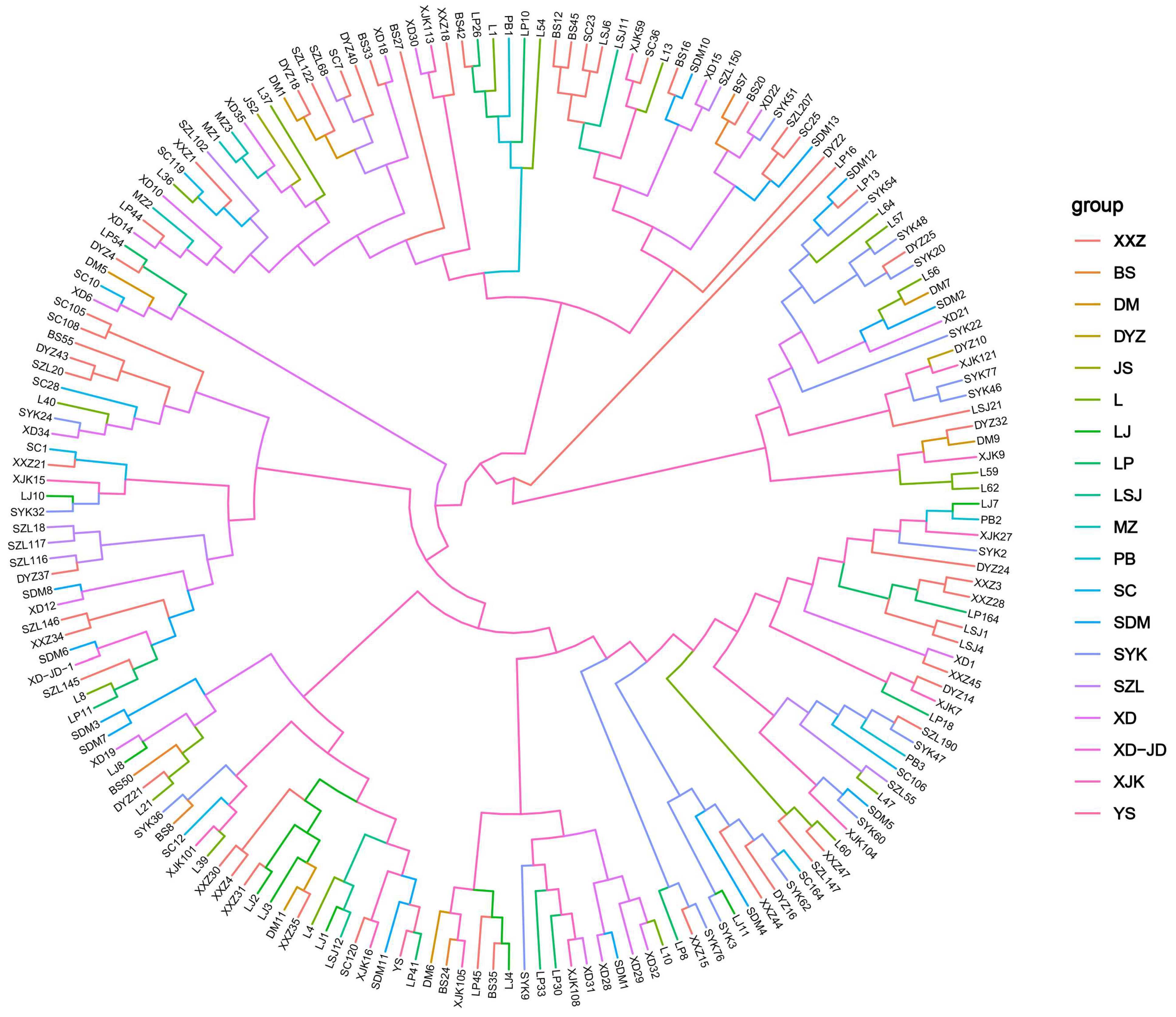
3.6. Cluster Analysis
4. Discussion
4.1. The Biology of M. acerina and Occurrence Regularity of Round Spot of P. notoginseng
4.2. Features of SSR Loci in M. acerina
4.3. Genetic Diversity of M. acerina in Yunnan
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Duan, L.; Xiong, X.J.; Hu, J.Y.; Liu, Y.; Li, J.; Wang, J. Panax notoginseng saponins for treating coronary artery disease: A functional and mechanistic overview. Front. Pharmacol. 2017, 8, 702. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.Y.; Fan, Y.; Yu, Q.T.; Ge, Y.-Z.; Yan, C.P.; Alolga, R.N.; Li, P.; Ma, Z.H.; Qi, L.W. Integrated evaluation of malonyl ginsenosides, amino acids and polysaccharides in fresh and processed ginseng. J. Pharm. Anal. 2015, 107, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Su, L.J.; Ren, Y.C.; Chen, Z.; Ma, H.-F.; Zheng, F.; Li, F.; Zhang, Y.Y.; Gong, S.S.; Kou, J.P. Ginsenoside rb1 improves brain, lung, and intestinal barrier damage in middle cerebral artery occlusion/reperfusion(mcao/r) mice via the pparγ signaling pathway. J. Nat. Med. 2022, 8, 561–571. [Google Scholar]
- Xia, P.G.; Zhang, S.C.; Liang, Z.S.; Qi, Z.H. Research history and overview of chemical constituents of Panax notoginseng. Chin. Tradit. Herb. Drugs 2014, 45, 2564–2570. [Google Scholar]
- Long, Y.J.; Mao, Z.S.; Zhu, S.S.; Chen, Z.J.; Wei, F.G.; Yin, Z.B.; Zhu, Y.Y.; He, X.H. The pathogen of Panax notoginseng root rust spot. Mycosystema 2015, 34, 177–185. [Google Scholar]
- Ma, Y.N.; Chen, C.J.; Li, Q.Q.; Xu, F.R.; Cheng, Y.X.; Dong, X. Monitoring antifungal agents of Artemisia annua against Fusarium oxysporum and Fusarium solani, associated with Panax notoginseng root-rot disease. Molecules 2019, 24, 213. [Google Scholar] [CrossRef]
- Tang, B.F.; Li, X.; Pu, L.; Zhao, Q.; Cui, X.; Ge, F.; Liu, D. A pathogenesis-related protein 10 gene pnpr10–3 was involved in molecular interaction between Panax notoginseng and Fusarium solani. Australas. Plant Path. 2019, 48, 447–456. [Google Scholar] [CrossRef]
- Lu, N.; Chen, Y.J.; Lu, H.J.; Liu, Y.L. Biological characterization of round spot pathogen on Panax notoginseng. J. Yunnan Agric. Univ. 2015, 2, 193–195. [Google Scholar]
- He, C.; Wang, H.L.; Jin, X.; Jin, B.H.; Su, S.; Duan, Y.N.; He, X.H. Identification of Alternaria species associated with black spot disease on Panax ginseng in Yunnan and Guangxi. Acta Phytophys. Sin. 2020, 50, 246–250. [Google Scholar]
- Jiang, N.; Qin, L.Y.; Ye, Y.F. Research advances in diseases of Panax notoginseng. J. South. Agric. 2011, 2, 149–153. [Google Scholar]
- Li, Y.; Long, S.; Li, X. Occurrence of root rot of Panax notoginseng caused by Fusarium oxysporum in China. Int. J. Agric. Biol. 2018, 20, 2175–2180. [Google Scholar]
- Dai, L.; Xu, Y.L.; Long, Y.J.; Du, L.S.; Du, Y.L.; He, X.H. The growth characteristic of mycelia and conidia for Mycocentrospora acerina. J. Yunnan Agric. Univ. (Nat. Sci.) 2017, 32, 27–35. [Google Scholar]
- Wall, C.J.; Lewis, B.G. Infection of carrot plants by Mycocentrospora acerina. Trans. Br. Mycol. Soc. 1980, 74, 587–593. [Google Scholar] [CrossRef]
- Davies, W.P.; Lewis, B.G.; Day, J.R. Infection of stored carrot roots by Mycocentrospora acerina (hartig) Deighton. Trans. Br. Mycol. Soc. 1977, 77, 139–151. [Google Scholar] [CrossRef]
- Garfinkel, A.R.; Chastagner, G.A. Strategies to Address Emerging Fungal Diseases in Peony (Paeonia lactiflora) in the United States; Horticultural Science: Korbeek-Lo, Belgium, 2019; pp. 199–206. [Google Scholar]
- Ellis, M.B. Dematiaceous hyphomycetes in Britain. In Transactions of the British Mycological Society; Cambridge University Press: London, UK, 1974. [Google Scholar]
- Hermansen, A. Weeds as hosts of Mycocentrospora acerina. Ann. Appl. Biol. 2010, 121, 679–686. [Google Scholar] [CrossRef]
- Wang, C.W.; Fu, J.F.; Wang, W.K. Brief report on the study of a new disease- the leaf withering disease of Asarum. J. Shenyang Agric. Univ. 1992, 23, 355–356. [Google Scholar]
- Chen, K.; Chen, S.X.; Yu, Z.W. A new disease on sanqi. Plant Prot. 1997, 1, 49. [Google Scholar]
- Cumagun, C. Population genetic analysis of plant pathogenic fungi with emphasis on Fusarium species. Philipp. Agric. Sci. 2007, 90, 244–256. [Google Scholar]
- Tsedaley, B.A. Review on Disease Detection, Pathogen Identification and Population Genetics in Fungi. J. Biol. Agric. Healthc. 2015, 5, 6–20. [Google Scholar]
- Salama, N.; Guillemin, K.; Mcdaniel, T.K.; Sherlock, G.; Tompkins, L.; Falkow, S. Whole-genome microarray reveals genetic diversity among Helicobacter pylori strains. Proc. Natl. Acad. Sci. USA 2000, 97, 14668–14673. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Ren, Z.H.; Liu, P.K.; Zhou, H.; Wang, H.H.; Liu, E.M. Analysis of genetic diversity of Magnaporthe oryzae in rice blast nursery in Taojiang of Hunan by SSR markers. Southwest China J. Agric. Sci. 2018, 31, 725–730. [Google Scholar]
- Sreenivasaprasad, S.; Talhinhas, P. Genotypic and phenotypic diversity in Colletotrichum acutatum, a cosmopolitan pathogen causing anthracnose on a wide range of hosts. Mol. Plant Pathol. 2010, 6, 361–378. [Google Scholar] [CrossRef] [PubMed]
- Barus, H.; Bayu, E.; Hanafiah, D.S. Identification genetic of soybean mutant (Glycine max L. Merril) based on fatty acid characters using simple sequence repeat (SSR) markers. In Proceedings of the 3rd International Conference Community Research and Service Engagements 2019, North Sumatra, Indonesia, 4 December 2019. [Google Scholar]
- Adjebeng-Danquah, J.; Manu-Aduening, J.; Asante, I.K.; Agyare, R.Y.; Gracen, V.; Offei, S.K. Genetic diversity and population structure analysis of Ghanaian and exotic cassava accessions using simple sequence repeat (SSR) markers. Heliyon 2020, 6, e03154. [Google Scholar] [CrossRef]
- Crauwels, S.; Zhu, B.; Steensels, J.; Busschaert, P.; Lievens, B. Assessing genetic diversity among Brettanomyces yeasts by DNA fingerprinting and whole-genome sequencing. Appl. Environ. Microbiol. 2014, 80, 4398–4413. [Google Scholar] [CrossRef] [PubMed]
- Colling, G.; Matthies, D. The effects of plant population size on the interactions between the endangered plant Scorzonera humilis, a specialised herbivore, and a phytopathogenic fungus. Oikos 2010, 105, 71–78. [Google Scholar] [CrossRef]
- Mcgrath, M.T.; Shishkoff, N. Resistance to Triadimefon and Benomyl: Dynamics and Impact on Managing Cucurbit Powdery Mildew. Plant Dis. 2001, 85, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Ortuño, D.; Pérez-García, A.; López-Ruiz, F.; Romero, D.; Vicente, A.D.; Torés, J.A. Occurrence and distribution of resistance to Qoi fungicides in populations of podosphaera fusca in south central Spain. Eur. J. Plant Pathol. 2006, 115, 215–222. [Google Scholar] [CrossRef]
- Wang, W.Y.; Zhao, C.L.; Chen, Z.J.; Wen, G.S.; Wei, F.G.; Long, T.G.; Li, S.W. Studies on the isolation, identification and in vitro growth rates of the three pathogenic fungi from Panax notoginseng cultivated in the Wenshan eparchy. Agric. Sci. Technol. 2015, 16, 1165. [Google Scholar]
- Li, H.; Li, J.; Xu, R.Y.; Song, F.S.; Li, L.; Wei, P.C.; Yang, J.B. Isolation of five rice non-endosperm tissue-expressed promoters and evaluation of their activities in transgenic rice. Plant Biotechnol. J. 2018, 16, 1138–1147. [Google Scholar] [CrossRef]
- Wang, Z.; Zheng, Q.M.; Cheng, W.T.; Mao, Y.Y.; Cai, Y.Q.; Ma, Y.H. Information analysis and molecular marker development of transcriptome SSR in Pitaya. Guizhou Agric. Sci. 2018, 46, 1–5. [Google Scholar]
- Wang, X.; Chen, L.; Zhao, C.L. Mining SSR Molecular Marker Sites with MISA Tool for Different Types of Sequences. Chin. Agric. Sci. Bull. 2016, 32, 150–156. [Google Scholar]
- Xu, D.L.; Chen, H.B.; Aci, M.; Pan, Y.H.; Shangguan, Y.N.; Ma, J.; Li, L.; Qian, G.; Wang, Q.X. De Novo assembly, characterization and development of EST-SSRs from Bletilla striata transcriptomes profiled throughout the whole growing period. PLoS ONE 2018, 13, e0205954. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.X.; Wei, A.Z.; Wang, L.H.; Liu, Y.L. Analysis of SSR loci and development of molecular markers in Zanthoxylum bungeanum transcriptome. J. Agric. Biotechnol. 2018, 26, 1226–1236. [Google Scholar]
- Azam, A.; Zahra, Z.; Zohreh, S.; Alireza, K.; Morteza, K.; Sirous, Z. Characterization and haplotype study of 6 novel STR markers related to the KCNQ1 gene in heterogeneous cardiovascular disorders in the Iranian population. Turk. J. Med. Sci. 2019, 49, 453–457. [Google Scholar]
- Zhou, X.J.; Wang, H.L.; Li, F.L.; Zhang, K.; Wang, Y.N.; Ya, H.Y. Development of polymorphic ssr markers in Rhododendron henanense subsp. lingbaoense based on rad-seq. J. Agric. Biotechnol. 2019, 27, 55–62. [Google Scholar]
- Zeng, X.Q.; Wang, Y.L.; Xu, Q.J.; Yuan, H.J.; Zaxi, L.B.; Nima, Z.X. Assessment of genetic diversity in Tibetan hulless barley germplasm (Hordeum vulgare L. var. nudum hk.f.) by SSR primers. Tcrop 2013, 33, 260–267. [Google Scholar]
- Ei-Komy, H.M.; Saleh, A.A.; Molan, Y.Y. Molecular characterization of early blight disease resistant and susceptible potato cultivars using RAPD and SSR markers. Afr. J. Biotechnol. 2012, 11, 37–45. [Google Scholar] [CrossRef]
- Yu, Z.F.; Yan, X.W.; Zhang, Y.H.; Yang, F.; Zhang, G.F. Genetic diversity analysis of different age of a Dalian population of the Manila clam Ruditapes philippinarum by EST-SSR. Acta Ecol. Sin. 2012, 32, 4673–4681. [Google Scholar]
- Liu, R.; YANG, J.S. Genetic diversity of some Dendranthema spp. based on RAPD analysis. J. Agric. Univ. Heb. 2010, 33, 60–65+83. [Google Scholar]
- Gaikwad, A.B.; Behera, T.K.; Singh, A.K.; Chandel, D.; Karihaloo, J.L.; Staub, J.E. Amplified fragment length polymorphism analysis provides strategies for the improvement of bitter gourd (Momordica charantia L.). Acta Hortic. 2008, 871, 71–78. [Google Scholar] [CrossRef]
- Wang, Z.J.; Wu, W.; Hu, B.; Zhang, H.L.; Bai, X.; Zhao, J.J.; Zhang, L.; Yan, X.J. Molecular epidemiology of Aleutian mink disease virus in China. Virus Res. 2014, 184, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Louarn, S.; Nawrocki, A.; Edelenbos, M.; Dan, F.J.; Jensen, O.N.; Collinge, D.B.; Jensen, B. The influence of the fungal pathogen Mycocentrospora acerina on the proteome and polyacetylenes and 6-methoxymellein in organic and conventionally cultivated carrots (Daucus carota) during post-harvest storage. J. Proteom. 2012, 75, 962–977. [Google Scholar] [CrossRef]
- Luo, H.; Wang, X.; Zhan, G.; Wei, G.; Zhou, X.; Zhao, J.; Huang, L.; Kang, Z. Genome-Wide Analysis of Simple Sequence Repeats and Efficient Development of Polymorphic SSR Markers Based on Whole Genome Re-Sequencing of Multiple Isolates of the Wheat Stripe Rust Fungus. PLoS ONE 2015, 10, e0130362. [Google Scholar] [CrossRef]
- Sharapova, N.; McMullen, M.D.; Schultz, L.; Schroeder, S.; Sanchez-Villeda, H. Development and mapping of SSR markers for maize. Plant Mol. Biol. 2002, 48, 463–481. [Google Scholar] [CrossRef] [PubMed]
- Toth, G. Microsatellites in Different Eukaryotic Genomes: Survey and Analysis. Genome Res. 2000, 10, 967. [Google Scholar] [CrossRef] [PubMed]
- Velasco, R.; Zharkikh, A.; Troggio, M.; Cartwright, D.A.; Viola, R. A high-quality draft consensus sequence of the genome of a Heterozygous grapevine variety. PLoS ONE 2007, 2, 1326. [Google Scholar] [CrossRef]
- Sia, E.A.; Kokoska, R.J.; Dominska, M.; Greenwell, P.; Petes, T.D. Microsatellite instability in yeast: Dependence on repeat unit size and DNA mismatch repair genes. Mol. Cell. Biol. 1997, 17, 2851–2858. [Google Scholar] [CrossRef] [PubMed]
- Mccouch, S.R.; Teytelman, L.; Xu, Y.; Katarzyna, B.L.; Karen, C.; Mark, W.; Fu, B.Y.; Maghirang, R.; Li, Z.K.; Xing, Y.Z.; et al. Development and mapping of 2240 new SSR markers for rice (Oryza sativa L.) (supplement). DNA Res. 2002, 9, 257–279. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.G.; Ishii, T.; Temnykh, S.; Chen, X.; Lipovich, L.; Mccouch, S.R.; Park, W.D.; Ayres, N.; Cartinhour, S. Diversity of microsatellites derived from genomic libraries and GenBank sequences in rice (Oryza sativaL.). Theor. Appl. Genet. 2000, 100, 713–722. [Google Scholar] [CrossRef]
- Lu, X.W.; Brewbaker, J.L. Molecular mapping of QTLs conferring resistance to Sphacelotheca reiliana (kühn) clint. Maize Genet. Coop. Newsl. 1999, 73, 36. [Google Scholar]
- Liu, C.; Li, J.; Qin, G. Genome-wide distribution of simple sequence repeats in pomegranate and their application to the analysis of genetic diversity. Tree Genet. Genomes 2020, 16, 1–9. [Google Scholar] [CrossRef]
- Wei, H.L.; Edo, K. Evolutionary pressures on simple sequence repeats in prokaryotic coding regions. Nucleic Acids Res. 2011, 6, 6. [Google Scholar]
- Xie, W.G.; Zhang, X.Q.; Ma, X.; Peng, Y.; Huang, L.K. Genetic variation and relationship in orchard grass (Dactylis glomerata l.) germplasm detected by SSR markers. Hereditas 2009, 31, 654–662. [Google Scholar] [PubMed]
- Zhou, Y.Y.; Wang, L.Q.; Fu, Y.L.; Yang, L.Q. A review of research on biological genetic resources and traditional knowledge. J. Northeast Agric. Univ. 2017, 16, 60–70. [Google Scholar]
- Sun, T.; Kong, L.W.; Wang, M.M.; Peng, Y.; Hong, X.Y. Development and characterization of novel EST-microsatellites for the citrus red mite, Panonychus citri (Acari: Tetranychidae). Syst. Appl. Acarol. 2014, 14, 499. [Google Scholar]
- Sun, T.T.; Wang, D.W.; Chen, L.; Wang, W.F.; Gong, D.P.; Chen, Y.Q.; Sun, Y.H. Characteristics and distribution of SSR loci in the coding region of potato genome and functional analysis of their encoding proteins. Mol. Plant Breed. 2015, 13, 171–177. [Google Scholar]
- Liu, X.F.; Yuan, W.Y.; Liang, D.; Shi, X.W.; Ma, Z.H.; Tian, J. Population genetic structures of Puccinia striiformis f. sp.tritici in Yunnan and Guizhou province. J. Yunnan Agric. Univ. (Nat. Sci.) 2016, 49, 95–100. [Google Scholar]
- Iwano, M.; Lee, J.L.; Lee, C.Y. Distribution of pathogenic races and changes in virulence of rice blast fungus, pyricularia oryzae cav. in Yunnan province, China. Jarq 1990, 23, 241–248. [Google Scholar]
- Wu, H.; Liu, B.L.; Wang, W.H.; Shi, Y.M.; Wei, S.F.; Zhou, H. Genetic diversity analysis of male sterile lines for three-line hybrid rice mainly applied in Guangxi. J. South. Agric. 2015, 46, 550–554. [Google Scholar]
- Mao, Y.X.; Zhang, X.L.; Hu, Z.J.; Zhao, Y. Genetic diversity of Trachycarpus fortunei (Hook.) H. Wendl germplasm resources in Guizhou by SRAP molecular markers analysis. J. South. Agric. 2020, 51, 27–35. [Google Scholar]
- Li, Y.F.; Zhang, X.L.; Liao, X.G.; Zhang, D. Evaluation study of multiplex PCR assay for detection diarrheagenic Escherichia coli. Henan J. Prev. Med. 2014, 2, 40. [Google Scholar] [CrossRef]
- Ferrucho, R.L.; Ceresini, P.C.; Ramírez-Escobar, U.M.; Mcdonald, B.A.; Domínguez, C.G. The population genetic structure of rhizoctonia solani AG-3PT from potato in the Colombian Andes. Phytopathology 2013, 103, 862–869. [Google Scholar] [CrossRef]
- Evenhuis, A.; Verdam, B.; Zadoks, J.C. Splash dispersal of conidia of Mycocentrospora acerina in the field. Plant Pathol. 2010, 46, 459–469. [Google Scholar] [CrossRef]
- Mcdermott, J.M.; Mcdonald, B.A. Gene flow in plant pathosystems. Annu. Rev. Phytopathol. 2003, 31, 353–373. [Google Scholar] [CrossRef]
- Wang, H.L.; Wang, F.; Jin, B.H.; Zhang, H.; Yang, K.; Wang, W.P.; Yang, M.; Zhu, S.S.; He, X.H. Sensitivity and fitness analysis of round spot of Panax notoginsen Mycocentrospora acerina to azoxystrobin, prochloraz and difenoconazole. Chin. J. Pestic. Sci. 2019, 21, 273–278. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus (Accession Number) | Repeat Motif | Primer Sequence | Size (bp) | Expected Heterozygosity (He) | Polymorphic Information Content (PIC) | Annealing Temperature (Tm) |
|---|---|---|---|---|---|---|
| MP40 (OM824348) | (CTA)27 | F: CACATGCTCAGTCATTTGTGG | 243 | 0.681 | 0.599 | 56 |
| R: GGTGCAATCGGAAAGAATTG | ||||||
| MP42 (OM824349) | (TC)19 | F: AAGCGCACTTGCCTATTGAT | 225 | 0.609 | 0.53 | 58.4 |
| R: GGTGAGTGTTGCTGACGAAA | ||||||
| MP2 (OM824341) | (CAT)14 | F: CGTCCATCTTCCTCTTCACC | 200 | 0.71 | 0.622 | 56.4 |
| R: GCTCATGTTCGATGGATGTG | ||||||
| MP49 (OM824352) | (GA)25 | F: GGAAGGAAATCCAGGTGTGA | 200 | 0.594 | 0.505 | 56 |
| R: CCCACTTCCTGTTTGCTTGT | ||||||
| MP54 (OM824356) | (CAA)19 | F: GTTGTTGCCAGCAAGAGTGA | 199 | 0.623 | 0.552 | 58.4 |
| R: AACAACCCTGGCACTACTCG | ||||||
| MP55 (OM824357) | (TTGA)19 | F: TTCCTCTCCCTCTCCCTCTC | 214 | 0.623 | 0.552 | 59 |
| R: ATGCTGCAAGTCTGTTGACG | ||||||
| MP56 (OM850345) | (TAG)25 | F: TGTGTGTGTGTTGTTGTTGTTG | 228 | 0.87 | 0.812 | 59.5 |
| R: TGACAAGCAAGTAGATTTTTACGTTT | ||||||
| MP4 (OM824343) | (GACA)6 | F: AGGGTAGCTCAAAGCCACTG | 273 | 0.725 | 0.665 | 58.4 |
| R: CTTTCCAAGCTGAGGGTGAG | ||||||
| MP58 (OM824358) | (GA)25 | F: TCGTTTTTGGAGCGTTCTTT | 209 | 0.739 | 0.659 | 54.4 |
| R: TGGACGCACTCCTTCTTTTC | ||||||
| MP52 (OM824355) | (CCA)11 | F: GCTTCGGTGTCTGGAATCAT | 179 | 0.609 | 0.53 | 56.4 |
| R: AAACTTCAATGTCGCCAAGG | ||||||
| MP51 (OM824354) | (TTG)19 | F: CGTCTCTGTTATTGCTGCTTT | 159 | 0.826 | 0.76 | 55.7 |
| R: CGCACAACCAATGAGAAACA | ||||||
| MP13 (OM824344) | (AGT)12 | F: CACGTCACGGAGCAAGTAGA | 211 | 0.696 | 0.622 | 57.4 |
| R: TGATGAGGTCCAACGGAGAT | ||||||
| MP30 (OM824342) | (GT)13 | F: CATGTGCATTGCTGTGTTGT | 170 | 0.725 | 0.644 | 61.4 |
| R: CAGCGAGTGAATGGAAGTGA | ||||||
| MP50 (OM824353) | (CTGT)19 | F: GCTTTACTTTGCCCGTCTGT | 200 | 0.768 | 0.701 | 55.4 |
| R: TGCATCTCCTCACATCCATC | ||||||
| MP65 (OM824362) | (CA)22 | F: ACCTCCACACCTGCACCTAC | 245 | 0.609 | 0.53 | 59.5 |
| R: GCGGGCTTGTAGTCGTAGAG | ||||||
| MP68 (OM824363) | (CAT)7 | F: GGATATGCCTCACCATTTGC | 166 | 0.797 | 0.739 | 55.4 |
| R: ATATGGAAGGCCGCAGTGTA | ||||||
| MP83 (OM824364) | (CTT)12 (ATGA)10 | F: TGAGCAGGGGCCAAATACTA | 156 | 0.779 | 0.702 | 54.4 |
| R: TTAAATTCCCATCCCCATCC | ||||||
| MP36 (OM824346) | (CAT)17 | F: ATCTGTCACCACCATCACCA | 193 | 0.87 | 0.812 | 59 |
| R: AGCTCGCGATCTAAACATCC | ||||||
| MP39 (OM824347) | (AGTG)24 | F: ATGTGTGTGTGTGCCTGGAT | 247 | 0.594 | 0.505 | 60 |
| R: TATATGCCCATTCCCATTCC | ||||||
| MP46 (OM824350) | (CACT)10 | F: TTCCTCTGACGCATCCTCTT | 207 | 0.638 | 0.535 | 60 |
| R: TGGGCATGTAATGAGTGGTG | ||||||
| MP47 (OM824351) | (CAGG)7 | F: GATTGTAAGCCGCAGAAGGT | 247 | 0.754 | 0.68 | 60 |
| R: TCACGACTCCATCACTCCAA | ||||||
| MP20 (OM824345) | (TACA)11 | F: TGTGTCGCTCACTCACTCAA | 239 | 0.754 | 0.671 | 59 |
| R: GGAAGGAGTGGAGTTGATGG | ||||||
| MP90 (OM824366) | (TC)18 | F: TCAAAACCGAAACCCAGAAA | 191 | 0.551 | 0.503 | 55.4 |
| R: GGGAGAAGAAGGGCAGAGG | ||||||
| MP108 (OM824368) | (TCG)10 (TCA)5 | F: TCACTACCCCTACCCCCTTT | 237 | 0.681 | 0.599 | 57.4 |
| R: CGGTCGGCATAGGGTATTTA | ||||||
| MP92 (OM824367) | (CTA)31 | F: ACCCCAACACTCAATCATCC | 219 | 0.71 | 0.643 | 54.7 |
| R: TCTGGCAAGAAGAAGAAATGC | ||||||
| MP62 (OM824360) | (CTA)20 | F: CAGAAAATCCTAGCTACTGCTGCT | 174 | 0.725 | 0.644 | 56.5 |
| R: TGCAGTCTCTTCACCCTGTTT | ||||||
| MP84 (OM824365) | (AGA)18 | F: TTCAATCGTGCAAGGTGTGT | 167 | 0.739 | 0.686 | 58 |
| R: GAGAGGAGCAGGGCATGTAG | ||||||
| MP115 (OM824371) | (AC)7(TC)12 | F: TCTGCTGCCATGTAGTGCTC | 246 | 0.768 | 0.692 | 55.4 |
| R: ATGTGATTTTGGGGGAAACA | ||||||
| MP63 (OM824361) | (TC)21 | F: CAGACTTCCCAGTCACCACA | 195 | 0.797 | 0.726 | 55.5 |
| R: TTGGCTACTACTGCACCAAAAA | ||||||
| MP113 (OM824369) | (TG)7(AG)10 | F: CATCTCTCATCTCCCCAGGA | 225 | 0.812 | 0.746 | 57.4 |
| R: AATCCCATCACACGCTTCTC | ||||||
| MP114 (OM824370) | (CTC)9 (TTC)8 | F: GATGTGCAGAGTTTCGGTCA | 232 | 0.913 | 0.862 | 55.4 |
| R: GGAAGCTGATTCATCCCAGT | ||||||
| MP61 (OM824359) | (CA)36 | F: TGGTGGCTAGTTGGTTGGAT | 212 | 0.928 | 0.878 | 56.4 |
| R: GGTCGTCACTGTTGCTTGAA |
| Population | Na | Ne | Nei’s Genetic Diversity | Shannon’s Information Index |
|---|---|---|---|---|
| Qujing | 1.6892 | 1.1108 | 0.0888 | 0.1658 |
| Honghe | 1.8041 | 1.1089 | 0.0896 | 0.1712 |
| Kunming | 1.7297 | 1.1103 | 0.0891 | 0.1674 |
| Puer | 1.6892 | 1.1099 | 0.0893 | 0.1676 |
| Lijiang | 1.4122 | 1.1187 | 0.0842 | 0.143 |
| Wenshan | 1.777 | 1.1099 | 0.0893 | 0.1693 |
| popID | Honghe | Lijiang | Puer | Kunming | Wenshan | Qujing |
|---|---|---|---|---|---|---|
| Honghe | 0.9938 | 0.9976 | 0.9988 | 0.9982 | 0.9977 | |
| Lijiang | 0.0062 | 0.9931 | 0.9937 | 0.9959 | 0.9946 | |
| Puer | 0.0024 | 0.007 | 0.9975 | 0.998 | 0.9978 | |
| Kunming | 0.0012 | 0.0064 | 0.0025 | 0.9979 | 0.9975 | |
| Wenshan | 0.0018 | 0.0041 | 0.002 | 0.0021 | 0.9983 | |
| Qujing | 0.0024 | 0.0054 | 0.0022 | 0.0025 | 0.0017 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Yang, K.; Huang, H.; Guo, L.; He, X. Development of Simple Sequence Repeat Markers and Genetic Diversity Evaluation of Mycocentrospora acerina in Yunnan Province, China. J. Fungi 2023, 9, 944. https://doi.org/10.3390/jof9090944
Wang H, Yang K, Huang H, Guo L, He X. Development of Simple Sequence Repeat Markers and Genetic Diversity Evaluation of Mycocentrospora acerina in Yunnan Province, China. Journal of Fungi. 2023; 9(9):944. https://doi.org/10.3390/jof9090944
Chicago/Turabian StyleWang, Huiling, Kuan Yang, Hongping Huang, Liwei Guo, and Xiahong He. 2023. "Development of Simple Sequence Repeat Markers and Genetic Diversity Evaluation of Mycocentrospora acerina in Yunnan Province, China" Journal of Fungi 9, no. 9: 944. https://doi.org/10.3390/jof9090944