
Abstract
Two new hydroxycitric acid lactone derivatives named cinatrins D (1) and E (2), and a new γ-lactam, virgaricin B (3), along with a known similar compound, virgaricin (4), were isolated from a fermentation broth of the fungus Virgaria boninensis FKI-4958. The absolute stereo structures of the new compounds were established by a combination of spectroscopic methods and chemical modifications. All three newly discovered compounds possessed weak antibacterial activity.
Similar content being viewed by others
Introduction
The genus Virgaria is a rare soil-inhabiting fungus, in which only two speices (Virgaria nigra and Virgaria boninensis) have been found so far. There have been few reports of chemical investigations of Virgaria species. Only one unusual diterpene, vinigrol has been reported to have been isolated from V. nigra.1 To the best of our knowledge, our previous chemical investigations of the species V. boninensis constituted the second report of its metabolites by the isolation of virgaricin (4) from a culture of the recently discovered V. boninensis FKI-4860.2, 3 In our continuing search for novel microbial metabolites from strains of V. boninensis, we investigated the chemical constituents of a fermentation broth of another strain of V. boninensis FKI-4958. As a result, we isolated and identified two new hydroxycitric acid lactone derivatives, cinatrin D (1) and cinatrin E (2), and a new γ-lactam, virgaricin B (3), together with virgaricin (4),2 which we have now renamed virgaricin A. The absolute configurations of 1–4 were elucidated using α-methoxy-α-(trifluoromethyl)phenylacetyl (MTPA) ester and phenylglycine methyl ester (PGME) as chiral anisotropic reagents.4, 5 In this paper, we report our reinvestigation of metabolites produced by differing strains of V. boninensis, which has led to the isolation of three additional new compounds 1–3, together with an initial appraisal of their bioactivity.
Results and discussion
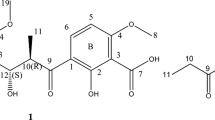
Compound 1 was obtained as a white amorphous solid and the molecular formula was elucidated as C18H30O7, with four degrees of unsaturation, by high resolution FAB-MS (HR-FAB-MS) (m/z 359.2072 [M+H]+ (calcd for C18H31O7, 359.2070)). Absorption at 3400 and 1749 cm−1 in the IR spectrum (KBr) suggested the presence of hydroxyl and carbonyl groups, respectively. The 1H and 13C NMR spectra of 1 are summarized in Table 1. The 1H and 13C NMR and HSQC spectra indicated the presence of one methyl, 11 methylenes, one aliphatic methine, one oxygen-substituted methine and four quaternary carbons (one oxygen-bearing carbon and three carbonyls). By treatment with trimethylsilyldiazomethane, 1 gave a dimethyl ester 1a, indicating the presence of two free carboxylic acid groups. The above functionalities could account for three of the four degrees of unsaturation, suggesting a monocyclic structure in 1. HMBC correlations (Figure 1) between H-1/C-2, C-3 and 3-CO, H-3/C-1, C-2 and 3-CO, and H2-4/C-2 and 3-CO indicated the presence of a γ-lactone. The chemical shift for C-2 (δC 81.7) clearly indicated that the hydroxyl group was attached to the quaternary carbon at C-2. Furthermore, HMBC correlations from H-1 to 1-CO and from H-1 and H-3 to 2-CO revealed the positions of two carboxyl groups and the formation of a garcinia acid (hydroxycitric acid lactone) moiety. The above structural information, in conjunction with 1H–1H COSY correlations and the molecular formula, suggested a saturated alkyl side chain that was composed of 12 carbons, attached directly to the methine carbon at C-3.

Key HMBC (arrows) and 1H–1H COSY (bold lines) correlations for 1 and 2.
The relative stereochemistry of 1 was elucidated by the NOE experiments. In 1H NMR experiments, the key proton signal for the tertiary hydroxyl group could not be confirmed using common deuterated solvents, including acetone-d6, chloroform-d, dimethyl sulfoxide (DMSO)-d6, methanol-d4 and pyridine-d5. On the other hand, fortunately, it was observed in the dimethyl ester 1a in DMSO-d6. In 1a, the key NOE correlations between H-1/H-3, H-1/OH-2 (δH 6.90) and H-3/OH-2 showed that these protons were located on the same side of the molecule (Figure 2). Thus, the relative structure of 1 was elucidated as 1R*, 2R* and 3R*. Evaluations using phenylglycine methyl ester (PGME)5 were performed to establish the absolute configuration of 1 (Figure 3). Treatment of 1 with PGME gave the corresponding 1,2-diamides 1b and 1c. The observed chemical shift differences, Δδ values (δS−δR) suggested the R configuration at C-2 (Figure 3). Therefore, the absolute stereochemistry was deduced as 1R, 2R and 3R.

Selected NOE correlations for 1a and 2a.

Δδ values (δS−δR) of PGME amides of 1.
Compound 2 was obtained as a white amorphous solid and the molecular formula was elucidated as C18H28O7, with five degrees of unsaturation, by HR-FAB-MS (m/z 379.1732 [M+Na]+ (calcd for C18H28O7Na, 379.1733)). The 1H and 13C NMR and HSQC spectra indicated the presence of 11 methylenes, including one exomethylene, one aliphatic methine, one olefinic methine, one oxygen-substituted methine and four quaternary carbons (one oxygen-bearing carbon and three carbonyls). The 1H and 13C NMR spectra of 2 were almost identical with those of 1, except for the presence of the terminal alkene group in the side chain. Analysis of the 2D NMR (1H–1H COSY and HMBC) spectra of 2 (Figure 1) suggested that 2 has the same planar structure of a garcinia acid moiety in 1. Furthermore, the relative configurations in 2 are also the same as 1, as elucidated by their similar correlations in the NOE spectra of 2a (Figure 2). This was supported by the agreement of 13C NMR data (Table 1) between 1 and 2. In consequence, the structure of cinatrin E (2) must be represented by formula 2, including the same relative stereochemistry as cinatrin D (1).
Compound 3 was obtained as a white amorphous powder and the molecular formula was elucidated as C17H23NO5, with seven degrees of unsaturation, by HR-FAB-MS (m/z 322.1658 [M+H]+ (calcd for C17H24NO5, 322.1654)). IR absorption bands indicated the presence of hydroxyl (3433 cm−1) and carbonyl (1709 and 1666 cm−1) functional groups. The 1H and 13C NMR spectra of 3 are summarized in Table 1. The 1H and 13C NMR and HSQC spectra indicated the presence of one methyl, three methylenes, eight olefinic methines, two methines adjacent to the heteroatom and three quaternary carbons (one oxygen-bearing carbon and two carbonyls). The 1H and 13C NMR spectral features of 3 were quite similar to those of virgaricin A (4) except for the side chain. The only difference was the presence of a double bond at C-16 in 3, which was confirmed by the 1H–1H COSY and HMBC correlations (Figure 4). The relative stereochemistry of 3 was elucidated by the ROESY experiment. The ROESY correlations between OH-3/H-4, H-4/H2-6 and H-5/H-8 indicated that 3 had the same relative stereochemistry as 4. In our previous report,2 the absolute stereochemistry of 4 was still under investigation. The modified Mosher’s method4 was applied to elucidate the absolute configuration at C-4 in 3 and 4. Treatment of 3 and 4 with (R)-(–)- and (S)-(+)-α-methoxy-α-(trifluoromethyl)phenylacetyl chlorides (MTPA-Cl) in pyridine gave the corresponding (S)- and (R)- MTPA esters 3a, 3b, 4a and 4b, respectively. The observed chemical shift differences, Δδ values (δS−δR) (Figure 5) clearly defined the S configuration at C-4. Therefore, the absolute configurations at C-3, C-4 and C-5 of 3 and 4 were deduced as 3S, 4S and 5S, respectively.

Key HMBC (arrows) and 1H–1H COSY (bold lines) correlations for 3.

Δδ values (δS−δR) of MTPA esters of 3 and 4.
In addition, we have isolated four known compounds, 2,3-dihydro-2-hydroxy-2,4-dimethyl-5-trans-propenylfuran-3-one, 2-(hydroxylmethyl)-3-(1-hydroxypropyl)phenol, isoochracein and mellein as metabolites from the FKI-6621 strain. The structures of these known compounds were confirmed by comparison of their spectroscopic data with those reported in the literature.2, 6, 7, 8 Details of the fermentation and isolation of the known compounds from the culture broth of FKI-6621 strain are described in the Supplementary Information.
Antimicrobial assay of the pure 1–3 using an agar diffusion method was carried out against 13 microorganisms.6 All of these compounds were found to be inactive against Kocuria rhizophila KB 212 (ATCC 9341), Mycobacterium smegmatis KB 42 (ATCC 607), Staphylococcus aureus KB 210 (ATCC 6538p), Escherichia coli KB 176 (NIHJ JC-2), Pseudomonas aeruginosa KB 115 (IFO 3080), Xanthomonas campestris pv. oryzae KB 88, Candida albicans KF 1, Saccharomyces cerevisiae KF 237 (ATCC 9763), Aspergillus niger KF 103 (ATCC 6275), Mucor racemosus KF 223 (IFO 4581) and Pyricularia oryzae KF 180, even at 100 μg per 6 mm disc. Compounds 1 and 2 were shown to be weakly active against the bacteria Bacillus subtilis KB 211 (ATCC 6633) with inhibition zones of 7.3 and 7.2 mm, respectively, as well as against E. coli KB 213 (NIHJ), with inhibition zones of 10.9 and 14.7 mm, respectively, at 100 μg per 6 mm disc. Compound 3 displayed only weak antibacterial activity against E. coli KB 213 (NIHJ) with an inhibition zone of 7.8 mm at 30 μg per 6 mm disc. In addition, all the compounds were tested for cytotoxicity against Madin–Darby canine kidney cells, but no cytotoxicity was observed at 100 μg ml−1.
Chemical constituents of the fungus Virgaria have also previously been studied by Ando et al.,1 but only two compounds had been isolated so far, of which one is identified in our previous study.2 In the present study, chemical reinvestigation of V. boninensis FKI-4958 has resulted in the isolation of three additional new natural products, cinatrins D (1) and E (2), and virgaricin B (3). V. boninensis FKI-4860 strain produced 4 (but not 1, 2 and 3).2 In addition, chemical screening of an another strain, V. boninensis FKI-6621, led to the isolation of four known compounds, 2,3-dihydro-2-hydroxy-2,4-dimethyl-5-trans-propenylfuran-3-one,7 2-(hydroxylmethyl)-3-(1-hydroxypropyl)phenol,8 isoochracein9 and mellein,10 but the strain did not produce 1–4. It is interesting that the fungus Virgaria peculiarly produces different types of metabolites. These results affirm that the rare fungus Virgaria is a potential source of novel natural products, some with interesting structures, that could lead us to find further novel and useful compounds.
Experimental procedure
General experimental procedures
Optical rotations were measured on a JASCO DIP-1000 polarimeter (JASCO, Tokyo, Japan). UV spectra were recorded on a Hitachi U-2810 UV–visible spectrometer (Hitachi, Tokyo, Japan). IR spectra were recorded on a Horiba FT-710 IR spectrometer (Horiba, Kyoto, Japan) using KBr discs. NMR spectra were acquired on an Agilent Technologies (Santa Clara, CA, USA) UNITY 400 (400 MHz for 1H and 100 MHz for 13C), a JEOL ECA 500 (JEOL, Tokyo, Japan, 500 MHz for 1H and 125 MHz for 13C) and an Agilent Technologies INOVA 600 (600 MHz for 1H and 150 MHz for 13C). FAB-MS and ESIMS spectra were conducted on JEOL JMS-700 MStation and JEOL JMS-T100LP (JEOL). Analytical and preparative reversed-phase HPLC were executed on a Senshu Pak Pegasil ODS SP100 column (4.6 i.d. × 250 mm) and a Senshu Pak Pegasil ODS column (20 i.d. × 250 mm), respectively, in conjunction with a JASCO MD-1510 multiwavelength detector and an Alltech 3300 ELSD detector (Nicholasville, KY, USA). Commercially available Silica gel 60 (70–230 mesh, Merck, Darmstadt, Germany) was used for open column chromatography. Preparative TLC was performed with silica gel plate (PLC Silica gel 60 F254, 0.5 or 1.0 mm thick, Merck).
Fungal material
The fungal strains V. boninensis FKI-4958 and FKI-6621 were isolated from soil samples collected with roots of subtropical plants in Chichi-jima, the Bonin Islands, Tokyo, Japan, 22 June 2007 and Haha-jima, the Bonin Islands, 1 April 2011, respectively. These strains were deposited in the Japan Collection of Microorganisms (JCM), Wako, with the strain numbers JCM 18623 (FKI-4958) and JCM 18624 (FKI-6621). We also examined Ascovirgaria occulta ATCC MYA-1219 (ex-type; teleomorph of V. nigra) obtained from the American Type Culture Collection (ATCC), Manassas, USA for comparison of sequence distances with respect to our strains.
Identification of fungal cultures
The two strains were classified as members of the Virgaria genus based on olive gray colonies and solitary, reniform, light brown sympodial conidia.11 Genomic DNA of the strains was isolated using the PrepMan Ultra Sample Preparation Reagent (Applied Biosystems, Foster City, CA, USA), according to the manufacturer’s instructions. Amplification of 28S ribosomal RNA gene (rDNA) Domain1/Domain2 (D1/D2) region and rDNA internal transcribed spacer (ITS) region, including 5.8S rDNA, were performed using primers NL1 and NL412 and primers ITS1 and ITS4,13 respectively. PCR reactions were performed according to the QIAGEN Fast Cycling PCR Kit protocol (Qiagen, Valencia, CA, USA). Amplifications were performed in a PCR Verity 96-well thermal cycler (Applied Biosystems), programmed with denaturation at 95 °C for 5 min, followed by 35 cycles, consisting of denaturation at 96 °C for 5 s, primer annealing at 50 °C for 5 s, extension at 68 °C for 21 s and a final elongation step at 72 °C for 1 min. After amplification of the D1/D2 and the ITS region templates, excess primers and dNTPs were removed from the reaction mixture using a QIAquick, PCR DNA Purification Kit (Qiagen). The PCR products were sequenced directly in both directions using primers NL1, NL4, ITS1 and ITS4 using a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems). The cycle sequencing reaction mixture had a total reaction volume of 10 μl, and contained 2.5 μl of template DNA (10–15 ng μl−1), 2-μl BigDye terminator premix, 4-μl ultra pure sterile water and 0.5-μl primer (5 pmol μl−1). Reactions were run in a PCR thermal cycler, programmed with denaturation at 96 °C for 1 min, then by 25 cycles of denaturation at 96 °C for 10 s, followed by primer annealing at 50 °C for 5 s and extension at 60 °C for 4 min. Sequencing products were purified using a BigDye XTerminator Purification Kit (Applied Biosystems), and samples were analyzed on an ABI PRISM 3130 Genetic Analyzer (Applied Biosystems). Contigs were assembled using the forward and reverse sequences with the SeqMan Pro and SeqBuilder programs from the Lasergene 10 package (DNASTAR, Madison, WI, USA). The DNA sequences were deposited at the DNA Data Bank of Japan (DDBJ) with accession number AB740959 (D1/D2) and AB670710 (ITS) for FKI-4958 and AB740960 (D1/D2) and AB740956 (ITS) for FKI-6621. With respect to sequences distances determined by the MegAlign programs from the Lasergene 10 package, the strains FKI-4898 and FKI-6621 had 97% similarity with the D1/D2 sequences of A. occulta ATCC MYA-1219 (AB740966) and 90% similarity with the ITS sequences of ATCC MYA-1219 (AB740957), respectively. From the results of morphological characteristics and sequences distances, the producing strains FKI-4958 and FKI-6621 were identified as a novel species of Virgaria.
Fermentation, extraction and isolation
Strain FKI-4958 was grown and maintained on a LcA agar slant consisting of 0.1% glycerol, 0.08% KH2PO4, 0.02% K2HPO4, 0.02% MgSO4·7H2O, 0.02% KCl, 0.2% NaNO3, 0.02% yeast extract and 1.5% agar (adjusted to pH 6.0 before sterilization). A loop of spores of V. boninensis FKI-4958 was inoculated into 100 ml of seed medium consisting of 2.0% glucose, 0.2% yeast extract, 0.5% Polypepton (Nihon Pharmaceutical, Tokyo, Japan), 0.05% MgSO4·7H2O, 0.1% KH2PO4 and 0.1% agar (adjusted to pH 6.0 before sterilization), in a 500-ml Erlenmeyer flask. The inoculated tube was incubated on a rotary shaker (210 r.p.m.) at 27 °C for 3 days. For the production of 1–3, a 1-ml portion of the seed culture was transferred to each of 19 500-ml Erlenmeyer flasks containing 100 ml of the production medium, consisting of 3.0% sucrose, 3.0% soluble starch, 1.0% malt extract, 0.3% Ebios (Mitsubishi Tanabe Pharma, Osaka, Japan), 0.5% KH2PO4 and 0.05% MgSO4·7H2O (adjusted to pH 6.0 before sterilization). Fermentation was carried out statically at 22 °C for 14 days.
The whole culture broth (1.9 l) was subsequently added to an equal amount of ethanol and then filtered. The filtrate was concentrated, under reduced pressure, to remove the ethanol and then extracted with EtOAc. The organic layer was concentrated to dryness in vacuo to afford a crude extract (1.13 g). The EtOAc extract was chromatographed on a silica gel column using a chloroform/methanol gradient solvent system of increasing polarity, to yield four fractions (Fr. 1–4). A portion of Fr. 2 (58.0 mg) eluted with chloroform/methanol (9/1) was further purified by reversed-phase HPLC (Senshu Pak Pegasil ODS, 70% acetonitrile with 0.1% trifluoroacetic acid, flow rate 8 ml min−1, UV at 200 nm and ELSD (evaporative light scattering detectors) detections) to give 1 (19.0 mg) and 2 (4.4 mg). Fr. 3 (259 mg) eluted with chloroform/methanol (8/2) was subjected to reversed-phase HPLC (Senshu Pak Pegasil ODS) with 50% acetonitrile at 8 ml/min detected at UV 350 nm to give 3 (12.0 mg) and 4 (5.0 mg).
Cinatrin D (1): white amorphous solid; [α]28D +50.1 (c 0.1, MeOH); IR (KBr) νmax 3400, 2917, 2850, 2359, 1749, 1221, 1084 cm−1; 1H NMR (600 MHz, pyridine-d5) and 13C NMR (150 MHz, pyridine-d5), see Table 1; HR-FAB-MS m/z 359.2072 [M+H]+ (calcd for C18H31O7, 359.2070).
Cinatrin E (2): white amorphous solid; [α]28D +41.6 (c 0.1, MeOH); IR (KBr) νmax 2925, 2854, 2362, 1786, 1741, 1201, 1074, 908 cm−1; 1H NMR (600 MHz, pyridine-d5) and 13C NMR (150 MHz, pyridine-d5), see Table 1; HR-FAB-MS m/z 379.1732 [M+Na]+ (calcd for C18H28O7Na, 379.1733).
Virgaricin B (3): white amorphous powder; [α]28D −127.4 (c 0.1, MeOH); UV (MeOH) λmax (log ɛ) 220 (3.89), 332 (4.54) nm; IR (KBr) νmax 3433, 1709, 1666, 1014 cm−1; 1H NMR (400 MHz, DMSO-d6) and 13C NMR (100 MHz, DMSO-d6), see Table 1; HR-FAB-MS m/z 322.1658 [M+H]+ (calcd for C17H24NO5, 322.1654).
Methylation of 1
To a solution of 1 (1.5 mg, 4.18 μmol) in a mixture of benzene/MeOH (v/v 9/1, 89.8 μl) at room temperature was added a 2.0-M solution of trimethylsilyldiazomethane (10.5 μl, 20.90 μmol) in hexanes. The reaction mixture was stirred at room temperature for 10 min. Then the solvent was removed in vacuo to afford the corresponding methyl ester of 1 (1a, 1.2 mg, 3.10 μmol). Methyl ester of 1 (1a): 1H NMR (DMSO-d6, 400 MHz) δ 6.90 (1H, s, OH-2), 5.29 (1H, s, H-1), 3.74 (3H, s, COOCH3), 3.70 (3H, s, COOCH3), 2.77 (1H, dd, J=6.9, 6.9 Hz, H-3), 0.85 (3H, t, J=6.9 Hz, H-15); HR-ESI-MS m/z 409.2206 [M+Na]+ (calcd for C20H34O7Na, 409.2202).
Methylation of 2
According to the procedure of 1a, 2 (1.6 mg, 4.49 μmol) with 2.0 M solution of trimethylsilyldiazomethane (11.2 μl, 22.45 μmol) in hexanes was converted to the corresponding methyl ester of 2 (2a, 1.6 mg, 4.16 μmol). Methyl ester of 2 (2a): 1H NMR (DMSO-d6, 400 MHz) δ 6.88 (1H, s, OH-2), 5.79 (dddd, J=17.2, 10.3, 6.9, 6.9 Hz, H-14), 5.28 (1H, s, H-1), 4.99 (dd, J=17.2, 1.7 Hz, H-15a), 4.93 (dd, J=10.3, 1.7 Hz, H-15b), 3.74 (3H, s, COOCH3), 3.70 (3H, s, COOCH3), 2.77 (1H, dd, J=6.9, 6.9 Hz, H-3), 2.00 (1H, ddd, J=6.9, 6.9, 6.9 Hz, H-13); HR-ESI-MS m/z 407.2047 [M+Na]+ (calcd for C20H32O7Na, 407.2046).
Preparation of the (R)- and (S)-PGME amides of 1
To a solution of 1 (3.8 mg, 10.60 μmol) and (R)-(−)-2-phenylglycine methyl ester hydrochloride (PGME; 6.4 mg, 31.80 μmol) in DMF (212 μl) was added 1H-benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate (PyBop, 16.5 mg, 31.80 μmol), HOBt (1-hydroxybenzotriazole hydrate) (4.3 mg, 31.80 μmol), and 4-methylmorpholine (10.5 μl, 95.40 μmol) at room temperature. After the reaction mixture was stirred at room temperature for 14 h, the reaction mixture was diluted with EtOAc (5 ml). The resulting organic layer was washed with 5% HCl aqueous solution (3 ml x3), saturated NaHCO3 aqueous solution (3 ml x3), and brine (3 ml x3), dried over Na2SO4, and concentrated in vacuo. The resulting crude product was purified by flash column chromatography (CHCl3/MeOH=100/1) to afford (R)-PGME amide 1b (5.6 mg, 8.58 μmol) in 81% yield as a pale yellow amorphous. (R)-PGME amide (1b): 1H NMR (pyridine-d5, 500 MHz) δ 10.58 (1H, d, J=7.5 Hz, NH), 9.33 (1H, d, J=7.5 Hz, NH), 7.52–7.09 (10H, m, phenyl group protons), 6.13 (1H, d, J=7.5 Hz, CHNH), 5.96 (1H, d, J=7.5 Hz, CHNH), 5.85 (1H, s, H-1), 3.80 (1H, dd, J=6.9, 6.9 Hz, H-3), 3.59 (3H, s, OCH3), 3.53 (3H, s, OCH3), 2.18 (1H, m, H-4a), 2.09 (1H, m, H-4b), 1.73 (1H, m, H-5a), 1.56 (1H, m, H-5b), 0.85 (3H, t, J=6.9 Hz, H-15); HR-ESI-MS m/z 675.3253 [M+Na]+ (calcd for C36H48N2O9Na, 675.3258). According to the procedure of 1b, 1 (3.4 mg, 9.49 μmol) with (S)-PGME (5.7 mg, 28.47 μmol) in DMF (189 μl), PyBop (14.8 mg, 28.47 μmol), HOBt (3.8 mg, 28.47 μmol) and 4-methylmorpholine (9.4 μl, 85.41 μmol) was converted to the corresponding (S)-PGME amide 1c (3.9 mg, 5.97 μmol) in 63% yield as a pale yellow solid. (S)-PGME amide (1c): 1H NMR (pyridine-d5, 500 MHz) δ 10.64 (1H, d, J=7.5 Hz, NH), 9.34 (1H, d, J=7.5 Hz, NH), 7.63–7.24 (10H, m, phenyl group protons), 6.17 (1H, d, J=7.5 Hz, CHNH), 5.92 (1H, d, J=7.5 Hz, CHNH), 5.90 (1H, s, H-1), 3.74 (1H, dd, J=6.9, 6.9 Hz, H-3), 3.56 (3H, s, OCH3), 3.41 (3H, s, OCH3), 1.91 (1H, m, H-4a), 1.71 (1H, m, H-4b), 1.58 (1H, m, H-5a), 1.39 (1H, m, H-5b), 0.86 (3H, t, J=6.9 Hz, H-15); HR-ESI-MS m/z 675.3266 [M+Na]+ (calcd for C36H48N2O9Na, 675.3258).
Preparation of the (R)- and (S)-MTPA ester derivatives of 3
Compound 3 (0.8 mg) was dissolved in 200 μl of dichloromethane, and pyridine (200 μl), and (S)-(+)-α-methoxy-α-(trifluoromethyl)phenylacetyl chloride (MTPA-Cl, 10 μl) were then added in sequence. The reaction mixture was allowed to stand at room temperature for 12 h, checked with TLC to make sure that the reaction was complete, and 200 μl of MeOH was then added. After standing for 5 min, the solvent was evaporated to dryness and purified by reversed-phase HPLC (Senshu Pak Pegasil ODS, 80% acetonitrile, flow rate 7 ml min−1, UV at 350 nm detection) to yield (R)-MTPA ester 3a (2.0 mg). By the same procedure, the (S)-MTPA ester 3b (1.7 mg) was obtained from the reaction of 3 (0.8 mg) with (R)-(−)-MTPA-Cl (10 μl). (R)-MTPA ester (3a): 1H NMR (CD3OD, 500 MHz) δ 7.10 (1H, dd, J=14.9, 11.5 Hz, H-9), 6.62 (1H, dd, J=14.9, 10.3 Hz, H-11), 6.37 (1H, d, J=14.9 Hz, H-8), 6.22 (1H, dd, J=14.9, 10.3 Hz, H-12), 6.05 (1H, m, H-10), 6.04 (1H, m, H-13), 5.57 (1H, d, J=6.9 Hz, H-4), 5.46 (1H, m, H-16), 5.46 (1H, m, H-17), 4.71 (1H, brd, J=12.6 Hz, H-6a), 4.33 (1H, dd, J=12.6, 1.7 Hz, H-6b), 4.03 (1H, m, H-5), 2.24 (2H, m, H-14), 2.13 (2H, m, H-15), 1.64 (3H, d, J=5.2 Hz, H-18); HR-ESI-MS m/z 776.2270 [M+Na]+ (calcd for C37H37F6NO9Na, 776.2270). (S)-MTPA ester (3b): 1H NMR (CD3OD, 500 MHz) δ 7.24 (1H, dd, J=14.9, 11.5 Hz, H-9), 6.69 (1H, dd, J=14.9, 10.9 Hz, H-11), 6.65 (1H, d, J=14.9 Hz, H-8), 6.23 (1H, dd, J=14.9, 10.9 Hz, H-12), 6.21 (1H, dd, J=14.9, 11.5 Hz, H-10), 6.05 (1H, ddd, J=14.9, 7.5, 7.5 Hz, H-13), 5.46 (1H, m, H-16), 5.46 (1H, m, H-17), 5.45 (1H, d, J=7.2 Hz, H-4), 4.62 (1H, dd, J=12.6, 2.3 Hz, H-6a), 4.23 (1H, dd, J=12.6, 2.9 Hz, H-6b), 3.89 (1H, m, H-5), 2.24 (2H, m, H-14), 2.13 (2H, m, H-15), 1.64 (3H, d, J=5.2 Hz, H-18); HR-ESI-MS m/z 776.2277 [M+Na]+ (calcd for C37H37F6NO9Na, 776.2270).
Preparation of the (R)- and (S)-MTPA ester derivatives of 4
The (R)-MTPA ester (4a) and the (S)-MTPA ester (4b) of 4 were produced by following the same procedure applied to 3. (R)-MTPA ester (4a): 1H NMR (CD3OD, 500 MHz) δ 7.11 (1H, dd, J=14.9, 10.9 Hz, H-9), 6.63 (1H, dd, J=14.9, 10.9 Hz, H-11), 6.38 (1H, d, J=14.9 Hz, H-8), 6.22 (1H, dd, J=14.9, 10.9 Hz, H-12), 6.07 (1H, m, H-10), 6.05 (1H, m, H-13), 5.57 (1H, d, J=7.5 Hz, H-4), 4.71 (1H, brd, J=12.6 Hz, H-6a), 4.33 (1H, brd, J=12.6 Hz, H-6b), 4.03 (1H, m, H-5), 2.19 (2H, dt, J=7.5, 7.5 Hz, H-14), 1.46 (2H, m, H-15), 1.20–1.40 (4H, m, H-16 and H-17), 0.90 (3H, t, J=7.5 Hz, H-18); HR-ESI-MS m/z 778.2430 [M+Na]+ (calcd for C37H39F6NO9Na, 778.2427). (S)-MTPA ester (4b): 1H NMR (CD3OD, 500 MHz) δ 7.24 (1H, dd, J=14.9, 11.5 Hz, H-9), 6.70 (1H, dd, J=14.9, 10.9 Hz, H-11), 6.65 (1H, d, J=14.9 Hz, H-8), 6.24 (1H, dd, J=14.9, 10.9 Hz, H-12), 6.21 (1H, dd, J=14.9, 11.5 Hz, H-10), 6.07 (1H, ddd, J=14.9, 7.5, 7.5 Hz, H-13), 5.45 (1H, d, J=6.9 Hz, H-4), 4.62 (1H, dd, J=12.6, 2.3 Hz, H-6a), 4.23 (1H, dd, J=12.6, 2.9 Hz, H-6b), 3.89 (1H, ddd, J=6.9, 2.9, 2.3 Hz, H-5), 2.19 (2H, dt, J=6.9, 6.9 Hz, H-14), 1.46 (2H, m, H-15), 1.24–1.40 (4H, m, H-16 and H-17), 0.90 (3H, t, J=6.9 Hz, H-18); HR-ESI-MS m/z 778.2423 [M+Na]+ (calcd for C37H39F6NO9Na, 778.2427).
Antimicrobial assay
Assay was carried out using a paper disc agar diffusion method (6 mm, ADVANTEC) described previously.9 Streptomycin or kanamycin (100 μg ml−1) was applied as a positive control for the microbial strains.
Cytotoxicity assay
Madin–Darby canine kidney cells were seeded into 96-well microplates (100 μl of a 20 × 104 cells per ml suspension in culture medium) and exposed to tested compounds (100 μg ml−1) for 72 h at 37 °C under 5% CO2. Adherent cells were fixed for 10 min with 25% glutaraldehyde, stained with 0.5% crystal violet, and solubilized with 0.5% SDS, followed by measurement of absorbance at 595 nm.
References
Ando, T. et al. Virgarol, a novel antihypertensive and platelet aggregation inhibitory agent producedcby fungus, Virgaria nigra. J. Antibiot. 41, 25–30 (1988).
Ishii, T. et al. Virgaricin produced by Virgaria sp. FKI-4860. J. Antibiot. 65, 139–141 (2012).
Nonaka, K., Ishii, T., Shiomi, K., Ōmura, S . & Masuma, R Virgaria boninensis, a new hyphomycete (Xylariaceae) from soils in the Bonin Islands, Japan. Mycoscience 54, 394–399 (2013).
Ohtani, I., Kusumi, T., Kashman, Y. & Kakisawa, H. High-field FT NMR application of Mosher’s method. The absolute configurations of marine terpenoids. J. Am. Chem. Soc. 113, 4092–4096 (1991).
Yabuuchi, T. & Kusumi, T. Phenylglycine methyl ester, a useful tool for absolute configuration determination of various chiral carboxylic acids. J. Org. Chem. 65, 397–404 (2000).
Iwatsuki, M. et al. Guadinomines, Type III secretion system inhibitors, produced by Streptomyces sp. K01-0509. J. Antibiot. 61, 222–229 (2008).
Grove, J. F. Metabolic products of Stemphylium radicinum. Part IV. Minor products. J. Chem. Soc. 12, 2261–2263 (1971).
Weber, D. et al. Metabolites from endophytes of the medicinal plant Erythrina crista-galli. Z. Naturforsch 60, 467–477 (2005).
Kameoka, H., Miyazawa, M. & Haze, K. 3-ethyl-7-hydroxypthalide from Forsythia japonica. Phytochemistry 14, 1676–1677 (1975).
Hamada, Y., Hara, O., Kawai, A., Kohno, Y. & Shioiri, T. Efficient total synthesis of AI-77-B, a gastroprotective substance from Bacillus pumilus AI-77. Tetrahedron 47, 8635–8652 (1991).
Rogers, J. D. & Ju, Y. M. Ascovirgaria occulta gen. et sp. nov., Jumillera hawaiiensis sp. nov., and Lopadostoma hawaiianum sp. nov. from Hawaii. Can. J. Bot. 80, 478–481 (2002).
O’Donnell, K. The Fungal Holomorph: Mitotic, Meiotic and Pleomorphic Speciation in Fungal Systematics (eds Reynolds, D. R. & Taylor, J. W.) 225–233 CAB International, Wallingford, CT, USA, (1993).
White, T. J., Bruns, T., Lee, S. & Taylor, J. in Amplification and Direct Sequencing of Fungal Ribosomal RNA Genes for Phylogenetics (eds Innis, M. A., Gelfand, D. H., Sninsky, J. J. & White, T. J.) 315–322 Academic Press, San Diego, CA, USA, (1990).
Acknowledgements
We are grateful to Dr Kenichiro Nagai and Ms. Noriko Sato, School of Pharmacy, Kitasato University for measurements of MS and NMR spectra.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article

Cite this article
Ishii, T., Nonaka, K., Sugawara, A. et al. Cinatrins D and E, and virgaricin B, three novel compounds produced by a fungus, Virgaria boninensis FKI-4958. J Antibiot 68, 633–637 (2015). https://doi.org/10.1038/ja.2015.45
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2015.45
This article is cited by
-
Virgaricins C and D, new pramanicin analogs produced by Apiospora sp. FKI-8058
The Journal of Antibiotics (2024)
-
Tolyprolinol, a new dipeptide from Tolypocladium sp. FKI-7981
The Journal of Antibiotics (2018)
