
Abstract
Cultivation of Cordyceps militaris, a valuable medicinal and edible fungus, has dramatically increased in Vietnam since 2010. During industrial production, parasitic white molds were found to infect the mycelia and fruiting bodies of C. militaris causing significant quality and yield losses. Two different fungal strains were obtained from the mycelia and fruiting bodies of C. militaris in Danang mushroom farms and were characterized by morphological and multiple DNA markers analysis. The sequence alignment of ITS, LSU and rpb2 markers revealed that the pathogens are related to the type species Lecanicillium coprophilum and Calcarisporium cordycipiticola with more than 99% sequence identities. The growth characteristics and pathogenic activities of the two isolated species on their host C. militaris were also investigated. The phylogenetic analysis based on the ITS sequences showed that L. coprophilum WF2611 is closer to its host C. militaris than C. cordycipiticola NT1504. To our knowledge, this is the first worldwide report of C. militaris infected by L. coprophilum which would be an useful information on prevention and control of the disease and be helpful for the industrial cultivation of C. militaris.
Similar content being viewed by others

Introduction
Cordyceps militaris (L.) Fr., one of the most valued edible and medicinal fungi, has long been used as an herbal drug and tonic in China, Korea, Japan and other East Asian countries due to their high content of bioactive compounds beneficial to human health1, 2. The bioactive constituents and pharmaceutical properties of C. militaris have similar to the wild-type Ophiocordyceps sinensis (formerly known as Cordyceps sinensis) and are widely used as a substitute for O. sinensis in health supplements3,4,5. Compared to O. sinensis, the artificial cultivation of C. militaris was easier and successfully archived in the early 1980s6. In addition, C. militaris can easily grow and produce fruiting bodies in solid and liquid media with a variety of carbon and nitrogen sources1. The cultivation of C. militaris has been widespread in China, Japan, Korea, Thailand and Vietnam. It is estimated that the annual value of production of C. militaris in China only is approximately 10 billion CNY (about 1.57 billion $ US)7.
During the large-scale production of C. militaris in China, fungal diseases frequently occur. The pathogens were first identified as Calcarisporium cordycipiticola sp. nov., Tricothecium crotocinigenum, Fusarium sp., Schizophyllum commune, Trichoderma harzianum, Purpureocillium lilacinum, Ustilaginoidea virens, Clonostachys rosea, T. ovalisporum, Penicillium expansum, Aspergillus oryzae and A. niger8,9,10. Among them, C. cordycipiticola is the most serious pathogen causing infectious diseases on fruiting bodies of C. militaris. The morphological characteristics and pathogenic mechanism of C. cordycipiticola on its host have been reported in detail, however, little information on the prevention and control of the disease is suggested11, 12.
Recently, the artificial cultivation of C. militaris in Vietnam has dramatically increased and a similar situation of fungal diseases has also been recognized. Among them, pathogens with white cottony colonies were often found to infect the mycelia and fruiting bodies of C. militaris causing significant quality and yield losses. However, no report for infectious mycoparasite of C. militaris in Vietnam has been published yet. Therefore, the aims of this study were to isolate and characterize the mycoparasites infected with C. militaris in Vietnam. The fungal pathogens isolated in this study were identified by multiple DNA markers including two markers of nuclear ribosomal regions (ITS—Internal Transcribed Spacer and LSU—28S nuclear ribosomal large subunit rRNA gene) and one protein coding sequence (rpb2 gene coding for the second largest subunit of RNA polymerase II). The morphological characteristics and the pathogenic activities of the fungal pathogens were then identified. Furthermore, the genetic relationships between the parasites and their host C. militaris were also investigated.
Results
Strain isolation and morphological observation
In this study, two different parasitic fungi with white mycelia forming a cottony layer on mycelia and fruiting bodies of C. militaris were isolated and serially plated onto PDA-Petri dishes to get the pure strains named WF2611 and NT1504, respectively (Fig. 1). Colonies of strain WF2611 were creamy-white, cottony, light yellow that slowly grown on PDA and reached 10–13 mm diameter at 25 °C after 7 days (Fig. 1a,b). However, colonies of strain NT1504 were pure-white, cottony that grown faster but reached a smaller diameter of 3–5 mm at 25 °C after 7 days (Fig. 1c,d).

White mold pathogens on mycelia and fruiting bodies of C. militaris and colonies of isolated strains on PDA medium at 25 °C after 7 days. Strain WF2611 (a,b) and strain NT1504 (c,d).
The microscopic analyses showed the conidiophores and conidia of the two isolated strains (Fig. 2). Conidiophores of strain WF2611 arose from aerial hyphae and moderately branched. Conidia (2–5 × 1–2 µm) aggregated to the apex of the conidiophores with shapes ranging from ellipsoidal to falcate or fusiform with round ends (Fig. 2a–c). For strain NT1504, there has more conidia formed around conidiophores and the conidia have different shapes but most of them are ovoid or fusiform with a size of 2–4 × 1–1.5 µm (Fig. 2d–f).

Microscopic analyses of isolated fungal strain WF2611 (a–c) and NT1504 (d–f). Conidiophores with conidia (a,b,d,e); conidia (c,f). Bars: 5 µm.
The pathogenicity test revealed that white cottony colonies of both strains developed on all five inoculated at any stage of growth, while the controls remained symptomless. The pathogens first invaded the mycelia, then proliferated on the surface of fruiting bodies of C. militaris. Strain NT1504 invaded the host C. militaris faster than strain WF2611. After 15–30 days, the white mold pathogens covered completely the fruiting bodies and the colour of the surface of fruiting bodies turned into grey and died. The morphological characteristics of white mold pathogens on inoculated cultures were identical to strains observed on the original infected fruiting bodies.
Strain identification
In order to identify the fungal pathogens, we amplified the complete ITS region of approximately 700 bp containing ITS1-5.8S-ITS2 from the DNA genome of isolated strains by using the primer pairs ITS5-F/ITS4-R (Fig. 3). Fragments of approximately 1100 bp containing 28S rDNA (LSU) from both isolated strains were also amplified by the primer pairs LROR-F/LR6-R. Finally, the rpb2 gene of approximately 1200 bp from both strains were amplified by the primer pairs RPB2-5F/RPB2-7cR. Those PCR products were then purified and sequenced by one direction for the sequence analysis and species identification.

Electrophoresis of the PCR products containing the complete ITS region of strain NT1504 (1) and WF2611 (2) on 0.8% agarose gel. M: 1 kb DNA ladder.
The ITS, LSU and rpb2 sequences obtained from sequencing data in this study were aligned with reference sequences by Basic Local Alignment Search Tool (BLAST) in Genbank. Firstly, the ITS sequence alignment showed that strain WF2611 and NT1504 exhibited 99.82% and 99.43% similarity with Lecanicillium coprophilum CGMCC 3.18986 (NR_163303) and Calcarisporium cordycipiticola MFLUCC 15–0685 (NR_144864), respectively. Sequences of ITS region of both strains were then deposited to GenBank with the accession numbers of OQ625887 and OQ625889, respectively. Similarly, the LSU sequences of strain WF2611 and NT1504 showed 99.89% and 99.42% similarity with L. coprophilum CGMCC 3.18986 (NG_067818) and C. cordycipiticola CGMCC 3.17905 (NG_067538), respectively. The GenBank accession numbers for the LSU sequences of WF2611 and NT1504 strains were OR272054 and OR272052, respectively. Finally, the rpb2 sequence alignment showed that strain WF2611 exhibited 99.16% and 100% similarity in nucleotide and protein sequences with L. coprophilum TBS415 (MH177624), respectively (Fig. S1). Although the rpb2 nucleotide sequence of strain NT1504 exhibited 99.40% similarity with C. coprophilum CGMCC 3.17940 (KX442607), the protein sequence alignment showed a reading frame shift near the 3’-end of the sequence due to the insertion of one Adenine (A) at nucleotide position 958 (Fig. S2). The sequences of rpb2 gene from strain WF2611 and NT1504 were deposited on GenBank with the aceession numbers OR500227 and OR500228, respectively. Based on those multiple markers analysis, we introduced the newly isolated strains as Lecanicillium coprophilum WF2611 and Calcarisporium cordycipiticola NT1504.
Phylogenetic analysis
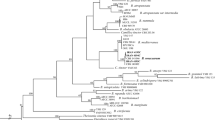
The ITS sequences from L. coprophilum WF2611, C. cordycipiticola NT1504 and related species downloaded from GenBank (Table 1) were aligned by Mega11 using the ClustalW algorithm. As shown in Fig. 4, the newly isolated strain C. cordycipiticola NT1504 formed a clade with Calcarisporium arbuscula CBS 900.68 and Mycophilomyces periconiae CPC 27,558 with bootstrap support of 71%. For the strain L. coprophilum WF2611, it formed a clade with Gamszarea microspora CGMCC 3.19313 and other Gamszarea species with bootstrap support of 83% and 99%, respectively. However, species of L. coprophilum formed a cluster distinct from other Lecanicillium species with strong bootstrap support of 97%. Furthermore, the phylogenetic tree in Fig. 4 also revealed that L. coprophilum WF2611 is phylogenetically closer to its host C. militaris than C. cordycipiticola NT1504.

Phylogenetic tree derived from Neighbour-joining analysis based on ITS sequences of the isolated strains and related taxa. Numbers at the branches represent bootstrap percentages. The newly isolated strains in this study were shown in bold.
Discussion
The large-scale prodution of C. militaris has been grown rapidly in Vietnam from 2010 and the problems of fungal diseases causing yield loss occurred very often, however, no information about the causative agents or the control methods have been reported until now. Therefore, our study was the first report of emerging fungal pathogens of C. militaris in Vietnam. Although there are many molecular markers used for barcoding, this study used ITS, LSU and rpb2 markers due to their advantages and their widely used for the identification of fungal samples. Firstly, the complete ITS region is commonly used for the classification of isolated fungal strains at the species level13. In addition, the available of 15.972 curated complete ITS sequences with correct taxonomic names of fungal type specimens from the ITS RefSeq Targeted Loci project provides great references for us to classify the isolates (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA177353/). Furthermore, the ITS5/ITS4 are also the standard primer pairs used to amplify the complete ITS region and there has 8.421 available ITS sequences amplified by these primer pairs in the GeneBank database13. The LSU sequence amplified by using the primer pairs LROR-F and LR6-R is normally used to identify fungi at higher taxonomic levels such as family or genera. In addition, the combination of LSU and ITS regions can also be valuable for the identification of fungi at the species level14. Using the ITS or LSU marker alone for fungal identification at species level might not suffice for some genera of phylum Ascomycota, including Aspergillus, Penicillium as well as Lecanicillium15. Therefore, in this study we used an protein-coding gene named rpb2 as an additional marker to deal with that problem. Protein-coding genes used for fungal identification have some advantages such as they occur as single copy in fungi and they have intron regions in their sequences, which sometimes evolve at a faster rate compared to ITS or LSU15. The nearly 100% similarity in ITS, LSU and rpb2 sequences of the isolated fungal samples and references from GenBank revealed that those markers are very suitable for the classification of microfungi at the species level.
In this study, two different fungicolous fungi were isolated from the fruiting bodies of infected C. militaris. Interestingly, one of them belongs to C. cordycipiticola that has firstly been isolated and recognized as an important fungal pathogen of C. militaris in China8. Biological characteristics and pathogenic mechanism of this fungicolous fungus on its host have been identified16. Furthermore, C. militaris is thought to be the only host of C. cordycipiticola11. In our work, the microscopic and growth characteristics of the newly isolated strain C. cordycipiticola NT1504 share similarities with previous reports of Sun et al.8. The pathogenicity test in this study also confirmed that C. cordycipiticola NT1504 is a strong fungal pathogen that can invade and reduce the production of C. militaris in a short period of time. The ability to produce a large number of conidia could be the reason for the rapid invasion of this pathogen16. Furthermore, the artificial cultivation of C. militaris in Vietnam must use air conditioners to maintain the suitable temperature for the growth of mushroom. This could disperse more fungal conidia into the air and thus accelerate the infection process.
The other fungal pathogen isolated in this work was identified as L. coprophilum. The species of Lecanicillium are recognized as mycoparasites of various arthropods, nematodes, and other fungi. Among them, L. coprophilum is first isolated and characterized from feces of Marmota monax, a species belonging to the group of large ground squirrels known as marmots. L. coprophilum differs from other Lecanicillium species by the morphological characteristics of conidiogenous cells, conidia, dictyochlamydospores and swollen hyphae17. Microscopic morphology of conidia and conidiophores of strain L. coprophilum WF2611 isolated in this study shares the same morphological characteristics with reports of Su et al.17. Many Lecanicillium species are entomopathogenic or fungicolous fungi, however, to our knowledge this is the first worldwide report that L. coprophilum is a mycoparasite of C. militaris.
The invasion process of a fungal parasite to its host depends on many factors. Among them, the close relationship between invader and its host could affect the effectiveness of the infection process. The phylogenetic analysis in this study indicated that L. coprophilum WF2611 is closer to its host C. militaris than C. cordycipiticola NT1504. This result is supported by the fact that the genera Lecanicillium and Cordyceps belong to the same family Cordycipitaceae, however, the genus Calcarisporium belongs to the family Calcarisporiaceae. In the order Hypocreales, Calcarisporiaceae and Cordycipitaceae are sister families18. The closer relationship between L. coprophilum WF2611 and its host C. militaris could be an advantageous characteristic for the effective interaction and invasion of the pathogen. Until now, no information about the pathogenicities or the infection mechanisms that L. coprophilum uses to invade the host C. militaris has been reported. Therefore, the pathogenicities, infection process, toxicities, host specificity, as well as the methods for prevention of this parasite should be further investigated.
Methods
Sample collection and culture conditions
Samples were collected from infected C. militaris growing on sterilized brown rice at Vinseed Biotechnology Co. Ltd. (N16o 01′ 37.64′′, E108o 22′ 06.95′′), Danang and some other farms in the central and southern regions of Vietnam in October 2022. Infected C. militaris fruiting bodies were aseptically transferred onto Potato Dextro Agar (PDA) in Petri dishes and incubated at 25 °C for 7 days. All samples were serially plated onto PDA in Petri dishes and incubated at 25 °C to get the pure strains. The ex-type living cultures were deposited in the Center for Pharmaceutical Biotechnology, School of Medicine and Pharmacy, Duy Tan University, Danang, Vietnam.
Morphological observation and pathogenicity test
All isolates were grown on PDA in Petri dishes and incubated at 25 °C for 7 days in darkness. The hyphal elongation was measured each day. Colony morphology and microscopic characteristics were examined, measured and photographed after incubation for 7 days. Microscopic observations were made from preparations mounted in 50% lactic acid. The structure and morphology of conidiophores were described from conidiophores obtained from the edge of conidiogenous pustules or fascicles of mature conidia.
The pathogenicity test was performed by gently dusting conidia of fungal pathogen onto five healthy fruiting bodies cultures of C. militaris and then growing under a condition of 25 °C, 85% humidity. Five noninoculated fruiting body cultures were used as controls9.
DNA extraction, PCR amplification, and DNA sequencing
Fresh mycelia (30 mg) were harvested from a 7-day-old plate and put into 1.5 mL Eppendorf tubes for genomic DNA extraction. Genomic DNA was extracted following the protocol of Plant genomic DNA extraction mini kit (Favorgen, Taiwan). In order to identify the fungal strains, three different molecular markers were amplified by polymerase chain reaction (PCR) and then sequenced. Firstly, the rDNA fragment containing ITS1-5.8S-ITS2 of approximately 700 bp was amplified using the primer pairs ITS5-F (5’-GGAAGTAAAGTCGTAACAAGG-3’) and ITS4-R (5’-TCCTCCGCTTATTGATATGC-3’)15. For the large subunit (LSU) ribosomal DNA (rDNA) marker based on the partial 28S rDNA, we used the primer pairs LROR-F (5’-ACCCGCTGAACTTAAGC-3’) and LR6-R (5’-CGCCAGTTCTGCTTACC-3’)15 to amplify a fragment of approximately 1100 bp. Lastly, the rpb2 gene (approximately 1200 bp in length) encoding the second largest subunit of RNA polymerase II was amplified using the primer pairs RPB2-5F (5’- GAYGAYMGWGATCAYTTYGG-3’) and RPB2-7cR (5’- CCCATRGCTTGYTTRCCCAT-3’)19. Each amplification reaction was performed in 25 µL reaction volume containing 2.0 µL of genomic DNA solution, 1.0 µL of each forward and reverse primers (100 pM/µL), 12.5 µL of 2 × DreamTaq PCR Master Mix (Thermo Fisher Scientific Baltics UAB, Lithuania) and 8.5 µL of ddH2O. The PCR parameters: 95 °C for 3 min; followed by 30 cycles at 95 °C for 30 s, 56 °C for 1 min, 72 °C for 1 min; and a final extension at 72 °C for 10 min were applied for the ITS and rpb2 markers. For the LSU marker, the annealing temperature parameter was 52 °C. The PCR products were then run on 0.8% agarose gel for 30 min and the DNA bands of each markers were cut and purified by GeneJET gel extraction kit (Thermo Fisher Scientific Baltics UAB, Lithuania). The purified PCR products were then sequenced by 1st BASE (Malaysia).
Phylogenetic analysis
The nucleotide sequences obtained from sequencing were compared to the references of ITS sequences from type material retrieved from BLAST database for ITS RefSeq at BioProject page (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA177353/) (NCBI, USA). The ITS sequence of isolates and related species from GenBank (Table 1) were aligned using ClustalW method. The phylogenetic tree was reconstructed using the neighbor-joining method and the reliability of the tree was estimated by the bootstrap method. All the evolutionary analyses were conducted in MEGA 1120.
Data availability
The DNA marker sequences of L. coprophilum WF2611 and C. cordycipiticola NT1504 are available on GenBank databases with the GenBank accession numbers: OQ625887 and OQ625889 for the ITS marker; OR272054 and OR272052 for the LSU marker; OR500227 and OR500228 for the rpb2 marker, respectively. Other data and analyses generated during the current study are included in this published article and in the supplementary information.
References
Cui, J. D. Biotechnological production and applications of Cordyceps militaris, a valued traditional Chinese medicine. Crit. Rev. Biotechnol. 35, 475–484 (2015).
Muslim, N. & Rahman, H. A possible new record of Cordyceps species from Ginseng Camp, Maliau Basin, Sabah, Malaysia. J. Trop. Biol. Conserv. 6, 39–41 (2010).
Das, S. K., Masuda, M., Sakurai, A. & Sakakibara, M. Medicinal uses of the mushroom Cordyceps militaris: Current state and prospects. Fitoterapia 81, 961–968 (2010).
Yue, K., Ye, M., Zhou, Z., Sun, W. & Lin, X. The genus Cordyceps: A chemical and pharmacological review. J. Pharm. Pharmacol. 65, 474–493 (2012).
Zhou, X., Gong, Z., Su, Y., Lin, J. & Tang, K. Cordyceps fungi: Natural products, pharmacological functions and developmental products. J. Pharm. Pharmacol. 61, 279–291 (2009).
Gu, H. S. & Liang, M. Y. Study on the manual cultivation of Cordyceps militaris. Med. Inf. Lett. 5, 51–52 (1987).
Dong, C. H. et al. Cordyceps industry in China: Current status, challenges and perspectives-Jinhu declaration for Cordyceps industry development. Mycosystema 35, 1–15 (2016).
Sun, J. Z., Dong, C. H., Liu, X. Z., Liu, J. K. & Hyde, K. D. Calcarisporium cordycipiticola sp. nov., an important fungal pathogen of Cordyceps militaris. Phytotaxa. 268, 135–44 (2016).
Liu, Q. & Dong, C. First report of white mold caused by Trichothecium crotocinigenum on Cordyceps militaris in China. Plant Dis. 104, 3253. https://doi.org/10.1094/PDIS-05-20-1024-PDN (2020).
Liu, Q., Wang, F., Xu, F. X., Xu, Y. Y. & Dong, C. H. Pathogenic fungi of artificially cultivated Cordyceps militaris. Mycosystema 40, 2962–2980 (2021).
Liu, Q. et al. Infection process and genome assembly provide insights into the pathogenic mechanism of destructive mycoparasite Calcarisporium cordycipiticola with host specificity. J. Fungi 7, 918. https://doi.org/10.3390/jof7110918 (2021).
Liu, Q. & Dong, C. Dual transcriptomics reveals interspecific interactions between the mycoparasite Calcarisporium cordycipiticola and its host Cordyceps militaris. Microbiol. Spectr. 11, e0480022. https://doi.org/10.1128/spectrum.04800-22 (2023).
Toju, H., Tanabe, A. S., Yamamoto, S. & Sato, H. High-coverage ITS primers for the DNA-based identification of Ascomycetes and Basidiomycetes in environmental samples. PloS ONE 7, e40863. https://doi.org/10.1371/journal.pone.0040863 (2012).
Schoch, C. L. et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. PNAS 109, 6241–6246 (2012).
Raja, H. A., Miller, A. N., Pearce, C. J. & Oberlies, N. H. Fungal identification using molecular tools: A primer for the natural products research community. J. Nat. Prod. 80, 756–770 (2017).
Liu, Q., Wan, J. X., Zhang, Y. C. & Dong, C. H. Biological characterization of the fungicolous Calcarisporium cordycipiticola, a pathogen of Cordyceps militaris. Mycosystema 37, 1054–1062 (2018).
Su, L. et al. Lecanicillium coprophilum (Cordycipitaceae, Hypocreales), a new species of fungus from the feces of Marmota monax in China. Phytotaxa 387, 055–062 (2019).
Sun, J. Z. et al. Calcarisporium xylariicola sp. nov. and introduction of Calcarisporiaceae fam. nov. in Hypocreales. Mycol. Prog. 16, 433–445 (2017).
Song, J., Chen, J. J., Wang, M., Chen, Y. Y. & Cui, B. K. Phylogeny and biogeography of the remarkable genus Bondarzewia (Basidiomycota, Russulales). Sci. Rep. 6, 34568. https://doi.org/10.1038/srep34568 (2016).
Tamura, K., Stecher, G. & Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027 (2021).
Acknowledgements
This work was supported by Duy Tan University and Vinseed Biotechnology Ltd Company. We thank Dr. Nguyen Minh Ly (University of Science and Education, The University of Danang, Vietnam) for his support in microscopic analyses. We also thank Cao Quang Khanh Duc (Duy Tan University) for his support in sample preparation.
Author information
Authors and Affiliations
Contributions
T.T.N. designed experiments. T.T.N. analysed the data and wrote the manuscript. Strains were isolated and characterized by T.N.G.L. Strains were identified and the phylogenetic tree was conducted by T.H.N. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nguyen, T.T., Le, T.NG. & Nguyen, T.H. First report of emerging fungal pathogens of Cordyceps militaris in Vietnam. Sci Rep 13, 17669 (2023). https://doi.org/10.1038/s41598-023-43951-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-43951-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
